
Abstract
Neuropsychiatric systemic lupus erythematosus (NPSLE) is a severe and potentially life-threatening complication of systemic lupus erythematosus (SLE), particularly in pediatric populations, in whom central nervous system involvement is often more aggressive and associated with long-term neurocognitive sequelae. Diagnosis remains challenging due to heterogeneous clinical manifestations and the lack of specific biomarkers. Traditionally, management has relied on high-dose corticosteroids, intravenous cyclophosphamide, and plasma exchange for refractory cases. However, emerging evidence supports the potential role of targeted immunomodulatory therapies, including rituximab, belimumab, anifrolumab, and emerging B-cell–directed strategies, although pediatric NPSLE-specific evidence remains limited. Novel agents such as type I interferon inhibitors, JAK inhibitors, and BTK inhibitors are currently under investigation in both adult and pediatric populations, offering the promise of more precise and less toxic interventions. This review summarizes current diagnostic approaches, first- and second-line therapies, and recent advances in biologic and small-molecule therapies for pediatric NPSLE. We also highlight ongoing clinical trials and future directions, emphasizing the importance of early recognition, multidisciplinary management, and the development of pediatric-specific guidelines. As understanding of the immunopathogenesis of NPSLE evolves, personalized therapeutic strategies may substantially improve long-term neurological and developmental outcomes in affected children.
Keywords
Introduction
Neuropsychiatric systemic lupus erythematosus (NPSLE) is one of the most severe complications of systemic lupus erythematosus (SLE), characterized by involvement of the central and/or peripheral nervous system. 1 Clinical manifestations of NPSLE are highly diverse, encompassing cognitive dysfunction, seizures, psychosis, mood disorders, headache, aseptic meningitis, movement disorders, cerebrovascular disease, demyelinating syndromes, transverse myelitis, peripheral neuropathy, and autonomic dysfunction. 2 These symptoms not only impair neurological function but also significantly compromise the quality of life and hinder the cognitive development of affected individuals. 3 Childhood-onset or juvenile SLE is generally defined as SLE diagnosed before 18 years of age. The median age at diagnosis is typically in early adolescence, although prepubertal onset may occur and is often associated with more severe disease activity and greater organ involvement. 4 In the UK juvenile-onset SLE cohort, most patients with neuropsychiatric involvement were female, and the median age at neurological involvement was 14 years. 5 In addition, children with SLE commonly exhibit multi-organ involvement-including renal, cutaneous, cardiovascular, and hematologic systems-further complicating diagnosis and management. 6
Epidemiological data indicate considerable variation in the incidence of pediatric SLE, with reported annual rates ranging from approximately 2–5 cases per 100,000 children. Among pediatric patients with SLE, neuropsychiatric involvement is observed in roughly 25–51%, depending on cohort characteristics, attribution models, and definitions used.5,7,8 In some cohorts, neuropsychiatric manifestations approach or exceed half of affected children, with seizures, cognitive dysfunction, and psychosis among the more prevalent clinical features. 3 For example, European cohort studies have reported cognitive deficits in approximately 30–70% of children diagnosed with NPSLE, highlighting the long-term impact of central nervous system involvement on academic performance and psychosocial development. 9
The diagnosis of NPSLE remains a major clinical challenge. The heterogeneity of neuropsychiatric manifestations and their frequent overlap with infectious, metabolic, or primary psychiatric disorders complicate early and accurate diagnosis, particularly in pediatric patients.10,11 Although conventional immunosuppressive therapies—including corticosteroids, cyclophosphamide, and antimalarial agents such as hydroxychloroquine—constitute the backbone of current treatment strategies, clinical responses remain highly variable. 12 A substantial proportion of patients exhibit partial or refractory disease, underscoring the limitations of non-specific immunosuppression and the urgent need for more targeted and individualized therapeutic approaches.
In recent years, the understanding of the underlying mechanisms of NPSLE has led to the development of novel therapeutic strategies, including biologic agents and targeted therapies. Emerging therapies such as Belimumab, Anifrolumab, and B-cell targeting therapies have provided new options for patients with refractory NPSLE.13–15 Cellular immunotherapies, such as CAR-T cell technology, have also begun to show preliminary validation in the treatment of NPSLE, providing new possibilities for future treatment strategies.16,17
This review aims to summarize current treatment strategies for pediatric NPSLE, with particular emphasis on therapeutic advances over the past 5 years. We will discuss the latest research on biologics, targeted immunotherapies, and immunocellular treatments. By highlighting ongoing clinical trials and unresolved challenges, we seek to delineate future directions toward precision medicine and pediatric-specific treatment paradigms in this complex disorder.
Methods
This narrative review was conducted to summarize current evidence regarding the diagnosis and treatment of pediatric neuropsychiatric systemic lupus erythematosus, with particular emphasis on emerging immunomodulatory therapies. A literature search was performed in PubMed/MEDLINE, Web of Science, and ClinicalTrials.gov from inception to April 2026. Search terms included “pediatric systemic lupus erythematosus”, “juvenile systemic lupus erythematosus”, “neuropsychiatric lupus”, “NPSLE”, “lupus cerebritis”, “belimumab”, “anifrolumab”, “rituximab”, “obinutuzumab”, “JAK inhibitor”, “CAR-T”, “biologic therapy”, and “biomarker”. We prioritized pediatric studies, pediatric NPSLE cohorts, systematic reviews, clinical trials, and major international recommendations. Because randomized controlled trials specifically addressing pediatric NPSLE are scarce, adult NPSLE studies, systemic SLE trials, retrospective cohorts, case series, and mechanistic studies were also considered when clinically relevant. Throughout the review, we distinguish pediatric NPSLE-specific evidence from adult NPSLE data and systemic SLE evidence extrapolated to neuropsychiatric disease.
Pathophysiological mechanisms of neuropsychiatric lupus
As summarized in Figure 1, the pathogenesis of pediatric NPSLE can be conceptualized into four interrelated mechanistic domains: cell- and cytokine-mediated neuroinflammation, complement dysregulation, autoantibody-mediated neuronal injury, and blood–brain barrier/neurovascular disruption with downstream brain structural changes. These domains are not mutually exclusive; rather, they interact dynamically to promote CNS inflammation, neuronal dysfunction, and neuropsychiatric manifestations. Pathophysiological mechanisms underlying pediatric neuropsychiatric systemic lupus erythematosus (NPSLE).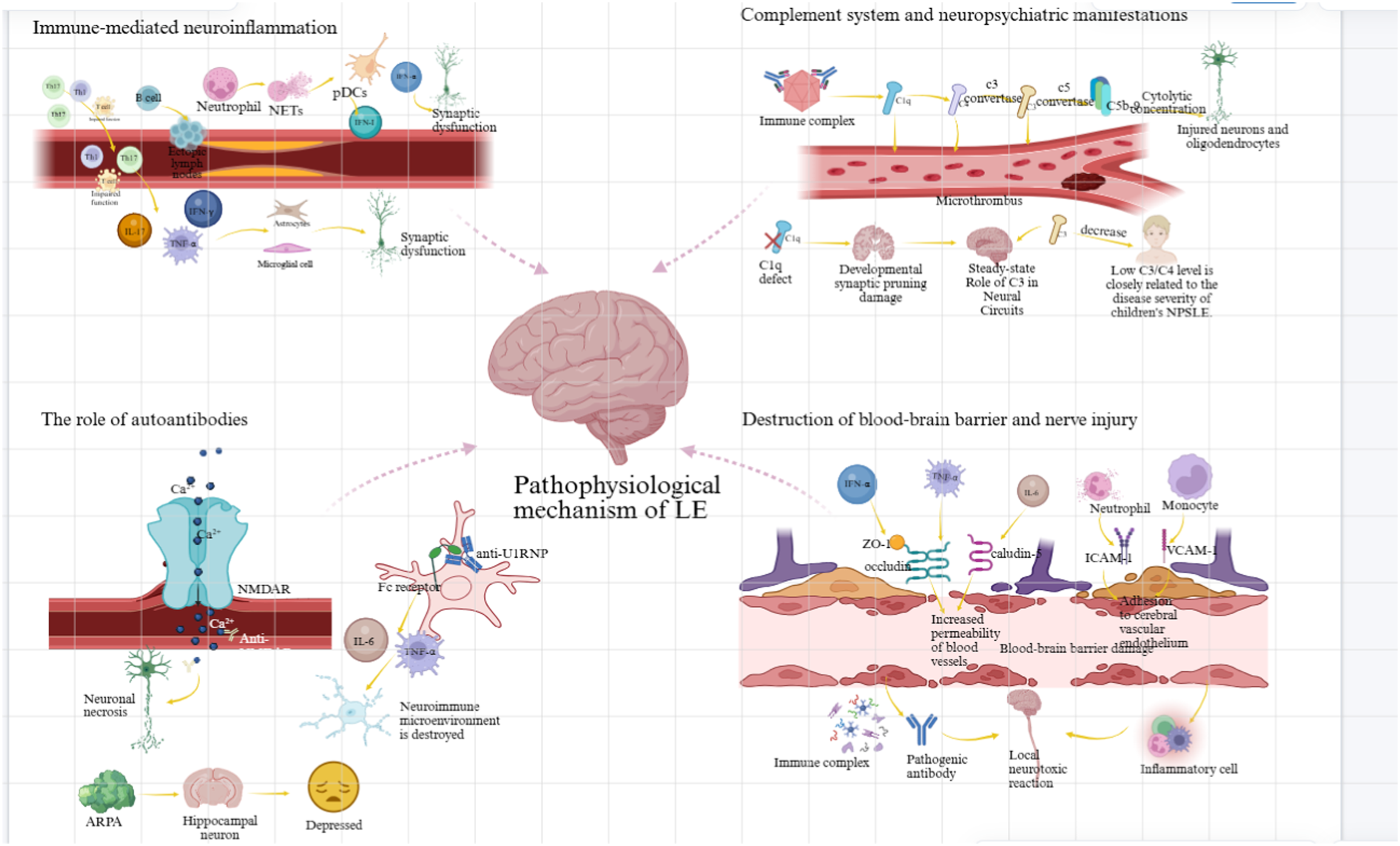
Cell- and cytokine-mediated neuroinflammation
A hallmark of NPSLE is chronic low-grade neuroinflammation driven by peripheral immune cell infiltration and local cytokine production within the CNS. 18 CD4+ T helper cells—particularly Th1 and Th17 subsets—are activated peripherally and migrate across the compromised BBB. Within the brain parenchyma, these cells secrete pro-inflammatory cytokines, including interferon-γ (IFN-γ), interleukin-17 (IL-17), and tumor necrosis factor-α (TNF-α), which activate microglia and astrocytes, leading to synaptic dysfunction and neuronal apoptosis. 19 Concurrently, impaired regulatory T cells (Tregs) fail to suppress this inflammatory cascade, thereby exacerbating immune dysregulation.20,21 B cells contribute not only through the production of pathogenic autoantibodies but also by forming ectopic lymphoid aggregates within the meninges, sustaining local immune activation. 22 In addition, neutrophils play an emerging role via the release of neutrophil extracellular traps (NETs), exposing nuclear antigens and activating plasmacytoid dendritic cells (pDCs). Activated pDCs produce large amounts of type I interferons (IFN-I), give rise to the characteristic “interferon signature” observed in both adult and pediatric NPSLE. 23 Labouret et al. reported that IFN-I activity was associated with pediatric NPSLE and may represent a promising biomarker of neuroinflammatory disease activity. 24 IFN-α can directly induce neuronal injury and upregulate endothelial adhesion molecules, thereby facilitating further leukocyte recruitment into the CNS.25,26
Complement system dysregulation and CNS homeostasis
Dysregulated complement activation plays a dual role in NPSLE. Excessive activation of the classical complement pathway leads to consumption of C1q, C3, and C4, promoting immune complex deposition within cerebral microvasculature, vasculitis, and microthrombotic injury.27,28 Additionally, assembly of the membrane attack complex (MAC; C5b–9) may directly damage neurons and oligodendrocytes at cytolytic concentrations, while subcytolytic levels can trigger intracellular signaling pathways that influence cell survival and inflammation. 29
Paradoxically, complement components also exert homeostatic functions within the CNS. Genetic C1q deficiency is a well-established risk factor for SLE, and insufficient complement activity within the brain may impair synaptic pruning during neurodevelopment, thereby contributing to neuropsychiatric dysfunction. 30 In a recent cohort of patients with hereditary C1q deficiency, enhanced type I interferon signaling and CNS inflammation were consistently observed, further supporting a link between complement dysfunction and neuroinflammation. 31 Consistent with this concept, lupus-prone murine models lacking C3 or C4 exhibit exacerbated cognitive deficits. 32 Clinically, hypocomplementemia-particularly reduced C3 and C4 levels—correlates with disease activity and severity in pediatric NPSLE.33,34
These observations underscore the dual and context-specific roles of complement in CNS homeostasis and injury, suggesting that therapeutic modulation of complement pathways will require careful patient selection and precise timing.
Autoantibodies and neuronal injury
A growing body of evidence implicates multiple autoantibodies in the direct or indirect mediation of CNS injury in NPSLE. Among these, anti–N-methyl-D-aspartate receptor (anti-NR2) antibodies—cross-reactive with anti–double-stranded DNA antibodies—bind to NR2 subunits on neuronal membranes, triggering excitotoxic calcium influx, mitochondrial dysfunction, and subsequent neuronal death. 35 In pediatric NPSLE, elevated anti-NR2 antibody titers have been associated with cognitive impairment and increased seizure frequency. 36 Anti-U1 ribonucleoprotein (U1RNP) antibodies are frequently detected in SLE patients with overlapping features of mixed connective tissue disease and have been linked to central nervous system involvement. Recent evidence suggests that anti-U1RNP immune complexes engage Fc receptors on microglia, promoting IL-6 and TNF-α secretion and disrupting the neuroimmune milieu. 37 Moreover, anti-ribosomal P antibodies have been associated with psychiatric manifestations, including depression and psychosis, potentially through direct interactions with hippocampal neurons. 38
Brain structural changes
Disruption of blood–brain barrier integrity is considered a critical early event in the pathogenesis of NPSLE. Pro-inflammatory cytokines—including TNF-α, IFN-α, and IL-6—downregulate tight junction proteins (e.g., occludin, claudin-5, ZO-1), increasing vascular permeability.39,40 In parallel, activated neutrophils and monocytes adhere to cerebrovascular endothelium through upregulation of adhesion molecules, including ICAM-1 and VCAM-1, further compromising BBB structure. 41 BBB disruption permits the entry of pathogenic autoantibodies, immune complexes, and inflammatory cells into the CNS, thereby initiating localized neurotoxic cascades. Neuroimaging studies using dynamic contrast-enhanced MRI have demonstrated that regions of BBB leakage spatially correspond to neuroimaging abnormalities in pediatric NPSLE. 42 Taken together, these findings highlight BBB dysfunction as both a pathogenic driver and a therapeutic barrier, emphasizing the need for treatment strategies capable of modulating neurovascular integrity and enhancing CNS drug delivery.
In summary, the pathophysiology of pediatric NPSLE reflects a complex interplay among immune cell activation, autoantibody-mediated neuronal injury, complement dysregulation, and BBB disruption. Recognition of these interconnected mechanisms provides a conceptual framework for the development of targeted and mechanism-driven therapies, moving beyond non-specific immunosuppression toward precision medicine approaches in pediatric neuropsychiatric lupus.
Conventional therapies in pediatric NPSLE
Traditional treatment approaches: From immunosuppression to symptom management
Pediatric NPSLE represents a severe and clinically challenging manifestation of SLE, encompassing diffuse or focal central nervous system dysfunction, including seizures, cognitive impairment, mood disturbances, psychosis, and encephalopathy. 43 Despite growing interest in targeted and biologic therapies, conventional immunosuppressive agents remain the cornerstone of initial management, particularly in acute and severe presentations. 44
Immunosuppressive therapy: Controlling inflammation and immune dysregulation
High-dose glucocorticoids remain the first-line therapy for acute inflammatory neuropsychiatric flares in pediatric SLE. In children, intravenous methylprednisolone pulse therapy is commonly administered at 10–30 mg/kg/day, with a maximum dose of 1 g/day, for 3–5 consecutive days, followed by an oral prednisone taper individualized according to disease severity, systemic involvement, and treatment response. Proposed mechanisms include inhibition of pro-inflammatory cytokines (such as IL-6 and TNF-α), stabilization of the blood–brain barrier, and reduction of autoantibody-mediated neuronal injury, although direct mechanistic evidence remains limited.45–47 Despite their efficacy in controlling acute disease activity, prolonged or repeated exposure to high-dose glucocorticoids is associated with significant adverse effects, including growth retardation, osteoporosis, hypertension, metabolic disturbances, and increased susceptibility to infection.48,49 These risks are particularly concerning in pediatric patients and underscore the importance of minimizing cumulative steroid exposure whenever possible. Importantly, glucocorticoids themselves may induce or aggravate neuropsychiatric symptoms, including mood disturbance, anxiety, insomnia, cognitive changes, and steroid-induced psychosis. This is particularly relevant in pediatric NPSLE, where disease-related psychiatric manifestations may be difficult to distinguish from treatment-related adverse effects. Careful temporal assessment, dose reduction when feasible, and collaboration with child psychiatry are therefore essential.
Cyclophosphamide (CYC) is generally reserved for severe, organ-threatening, or refractory inflammatory NPSLE. In pediatric practice, intravenous cyclophosphamide is often administered at approximately 500–750 mg/m2 or 700 mg/m2 per pulse, usually for 3–6 pulses, with a maximum of 1 g per pulse and careful attention to cumulative exposure, commonly aiming to remain below approximately 6 g when possible because of gonadotoxicity concerns. Through broad immunosuppressive effects—including depletion of autoreactive B cells and suppression of T-cell proliferation—CYC reduces immune complex deposition and neuroinflammatory activity. 50 Observational studies suggest that the combination of cyclophosphamide and corticosteroids may improve neurological outcomes in severe NPSLE; however, high-quality pediatric-specific prospective data are lacking. At the same time, concerns regarding gonadal toxicity, hemorrhagic cystitis, infertility, and secondary malignancies have prompted efforts to limit cumulative exposure and explore alternative induction strategies. 51 These limitations have been a major driver of interest in less toxic, targeted therapies.
Hydroxychloroquine (HCQ) is recommended as background therapy for all SLE patients, including children. Beyond its immunomodulatory effects—such as inhibition of Toll-like receptor 7/9 signaling and modulation of antigen presentation—HCQ may exert indirect neuroprotective effects by reducing systemic inflammation and thrombotic risk. 52 Observational studies in pediatric SLE cohorts have shown that regular HCQ use is associated with improved disease control and reduced organ damage accrual. While direct evidence linking HCQ to a reduced incidence of neuropsychiatric SLE is limited, consistent adherence to HCQ therapy correlates with fewer severe disease manifestations and hospitalizations. 53 However, HCQ demonstrates limited efficacy in treating active neuropsychiatric episodes. Furthermore, its therapeutic benefits may be compromised by pharmacogenetic variability (e.g., CYP2C8/CYP3A4 polymorphisms), leading to unpredictable drug exposure. 54 Given the risk of retinopathy associated with long-term use, regular ophthalmologic screening is mandatory. 55
Symptomatic management: Addressing neuropsychiatric manifestations
Symptom-directed therapies play a critical adjunctive role in the management of pediatric NPSLE and are essential for optimizing functional outcomes and quality of life. For seizure control, antiepileptic drugs (AEDs) such as levetiracetam, valproate, and lamotrigine are commonly prescribed. Levetiracetam is often preferred due to its favorable safety profile and minimal drug interactions. 44 Notably, enzyme-inducing AEDs like phenytoin should be avoided because of their potential to exacerbate lupus activity. 56
Psychiatric manifestations, including psychosis and mood disturbances, are typically managed with second-generation antipsychotics such as olanzapine or quetiapine, which are better tolerated in pediatric populations.57,58 However, metabolic side effects—including weight gain, insulin resistance, and hyperprolactinemia—require careful monitoring. 59 While no disease-modifying pharmacologic therapies are currently available for cognitive dysfunction, early cognitive rehabilitation, psychological support, and educational interventions may help preserve neurodevelopmental outcomes. 60
Challenges and limitations of conventional therapies
Despite their widespread use, conventional therapies for pediatric NPSLE are associated with several important limitations. Heterogeneity in treatment response suggests the existence of distinct pathophysiological subtypes that may be resistant to non-specific immunosuppression. 61 In addition, the delayed onset of action of many agents limits their effectiveness during acute, life-threatening neuropsychiatric crises. Long-term immunosuppression increases infection-related morbidity and mortality, especially in pediatric patients with compromised immune defenses. 62 Furthermore, most therapeutic recommendations are extrapolated from adult studies, and randomized controlled trials (RCTs) focused on pediatric NPSLE are exceedingly rare. 5
Long-term and mid-term outcome data further highlight the limitations of current treatment approaches. Cohort studies have shown that pediatric patients with NPSLE have poorer long-term outcomes than those without neuropsychiatric involvement, including greater cumulative damage, persistent neurocognitive or psychiatric sequelae, and impaired health-related quality of life.63,64 Recent follow-up data also suggest that although many mild neuropsychiatric manifestations may improve after treatment, residual symptoms can persist at 6 and 12 months in a subset of patients. 8 These findings indicate that management should not be limited to acute immunosuppression or symptomatic control, but should also include longitudinal neuropsychological assessment, mental health support, school-based accommodations, rehabilitation, and strategies to prevent cumulative damage.
In summary, while traditional therapies remain indispensable in the management of pediatric NPSLE, their limitations—including variable efficacy, cumulative toxicity, and lack of personalization—highlight the need for biomarker-driven strategies and novel therapeutics. 65 The future lies in transitioning from empirical treatment to precision medicine guided by immunophenotyping and neuroimaging biomarkers.
Advances in targeted therapies for pediatric neuropsychiatric lupus
Conventional therapies for SLE have long relied on glucocorticoids and cyclophosphamide. However, their use is constrained by significant long-term toxicities. Recent advances in elucidating disease pathogenesis—particularly the roles of B-cell hyperactivity, type I interferon (IFN-I) pathway activation, and broader immune dysregulation—have paved the way for more targeted biologic and cellular therapies (Figure 2). Therapeutic landscape of pediatric neuropsychiatric systemic lupus erythematosus (NPSLE): from conventional immunosuppression to targeted and cellular immunotherapies.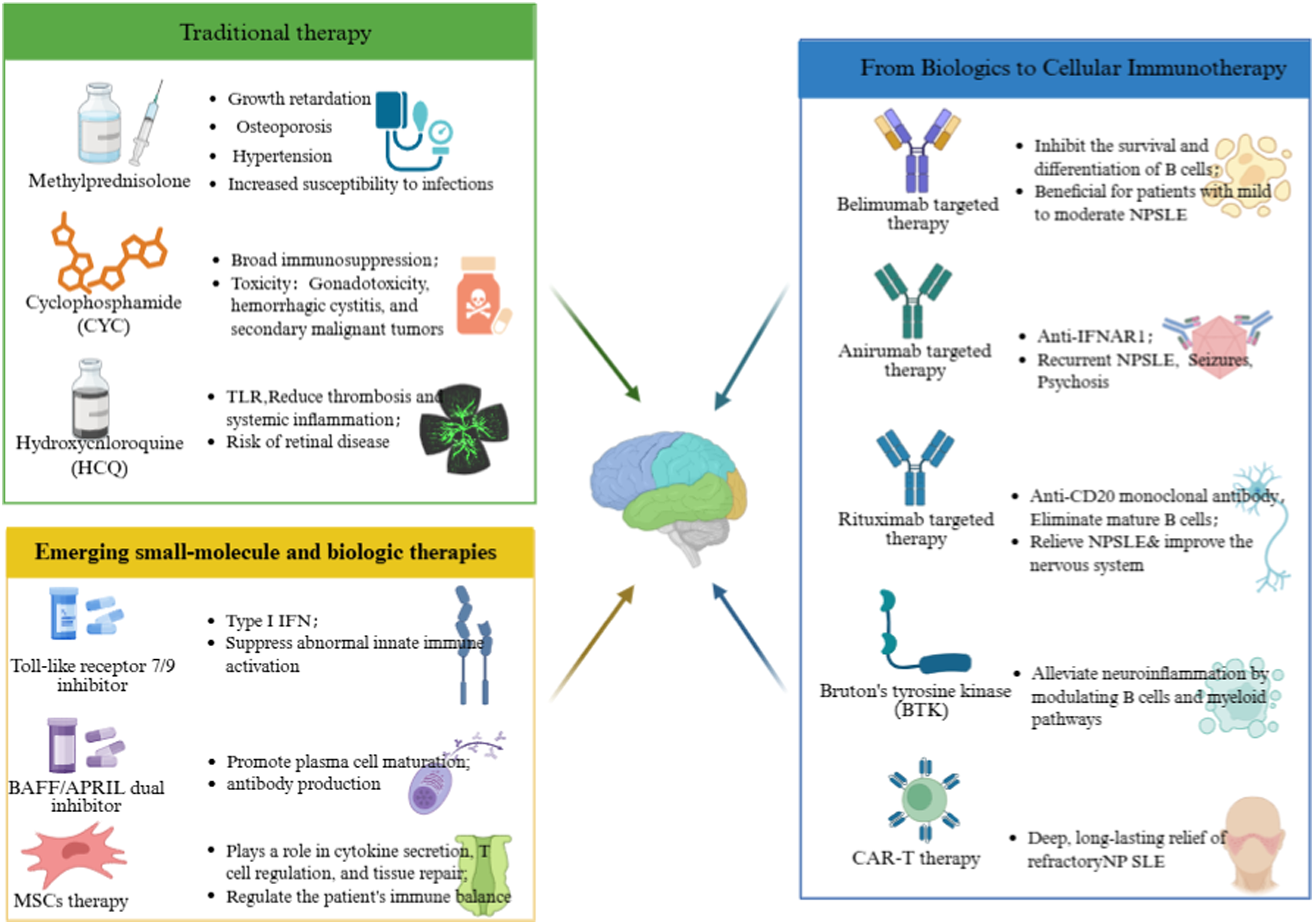
Because pediatric NPSLE-specific randomized trials are lacking, the evidence supporting targeted therapies varies substantially across agents. In this section, we explicitly distinguish evidence derived from pediatric NPSLE cohorts, adult NPSLE studies, systemic SLE trials without active CNS involvement, and mechanistic or preclinical studies. This distinction is critical because efficacy in systemic SLE cannot be assumed to translate directly into benefit for inflammatory CNS disease, particularly when blood–brain barrier penetration, neurovascular injury, and attribution of neuropsychiatric events differ across patients.
Belimumab in pediatric NPSLE
Belimumab is a fully human IgG1λ monoclonal antibody targeting B-lymphocyte stimulator, also known as BLyS or BAFF, thereby inhibiting B-cell survival and differentiation. Belimumab is approved for children aged ≥5 years with active SLE on the basis of pediatric pharmacokinetic, safety, and efficacy data, including the PLUTO trial. 66 Although belimumab is not specifically indicated for neuropsychiatric disease, accumulating evidence suggests potential benefit in selected patients with mild-to-moderate CNS involvement. Real-world SLE studies have reported reductions in systemic disease activity and steroid-sparing effects following belimumab initiation, 67 but these data should not be interpreted as direct evidence of efficacy in pediatric NPSLE. Available pediatric SLE data, including the PLUTO trial, have not identified major new safety signals; however, patients with active central nervous system involvement were generally excluded from randomized trials, limiting conclusions regarding CNS-specific safety and efficacy. 68
High-quality randomized controlled trial evidence for belimumab in pediatric SLE is provided by the PLUTO trial, a double-blind, placebo-controlled study evaluating intravenous belimumab in children with active childhood-onset SLE. 66 In this trial, belimumab plus standard therapy demonstrated numerically higher SRI4 response rates compared with placebo (52.8% vs 43.6%), although the difference did not reach statistical significance. Improvements were also observed in several secondary disease activity measures, including reductions in flare rates and cumulative prednisone dose. Safety profiles were comparable between treatment groups, with serious adverse events occurring in 17.0% of belimumab-treated patients versus 35.0% in the placebo group, and serious infections reported in 1.9% and 2.5% of patients, respectively. Notably, patients with active central nervous system involvement were excluded from the PLUTO trial, limiting the direct applicability of these findings to pediatric NPSLE.
Although randomized trials of belimumab have generally excluded patients with severe active CNS lupus, several real-world studies have provided NPSLE-relevant observations. Plüß et al. evaluated routinely administered belimumab in patients with SLE and reported clinical improvement not only in proteinuria but also in a subset of patients with neuropsychiatric lupus. 67 Although this study was retrospective and included mainly adult patients, it provides important real-world evidence that belimumab may contribute to stabilization or improvement of selected neuropsychiatric manifestations when used as add-on therapy. However, the small number of NPSLE patients, heterogeneous neuropsychiatric phenotypes, and absence of standardized neurocognitive outcome measures limit definitive conclusions.
Nevertheless, large-scale randomized controlled trials specifically evaluating belimumab in severe pediatric NPSLE are lacking. 44 Given its limited penetration across the blood-brain barrier, belimumab is thought to exert its effects primarily through peripheral B-cell modulation and cytokine regulation rather than direct CNS activity. 61 Consequently, belimumab should currently be regarded as a potential steroid-sparing option for selected patients with systemic B-cell–driven disease and mild-to-moderate neuropsychiatric involvement, rather than as a proven therapy for severe pediatric NPSLE.
Anifrolumab in pediatric NPSLE
Anifrolumab, a fully human IgG1κ monoclonal antibody targeting the type I interferon receptor 1 (IFNAR1), suppresses the interferon pathway—a hallmark of SLE pathogenesis present in over 80% of patients. 69 Phase III clinical trials in adults (TULIP-1 and TULIP-2) demonstrated significant improvements in disease activity, cutaneous and musculoskeletal manifestations, and corticosteroid-sparing outcomes. 70
Long-term extension data from anifrolumab trials have demonstrated sustained improvements in patient-reported outcomes and overall disease control in adult SLE. 71 However, these trials were not designed to evaluate active NPSLE, and CNS-specific efficacy remains uncertain. The ongoing BLOSSOM trial (NCT05835310), a phase II/III multicenter study enrolling children aged 5–17 years with active or refractory SLE, will provide critical data regarding safety, pharmacokinetics, and efficacy in pediatric populations. It should be noted that the BLOSSOM trial excludes patients with a history or evidence of suicidal ideation. This criterion is clinically important because mood disorders, depression, and suicidal ideation may occur in pediatric NPSLE. Therefore, trial results may underrepresent patients with severe psychiatric involvement, and extrapolation to the full pediatric NPSLE spectrum should be cautious.
Evidence for anifrolumab in NPSLE remains preliminary but is emerging. A recent real-world conference report described the use of anifrolumab in SLE patients with neurological manifestations, suggesting potential improvement in selected neuropsychiatric symptoms. 72 Another conference case report described anifrolumab use in lupus headache, with reported clinical improvement. 73 These observations are hypothesis-generating and should be interpreted cautiously because they are based on small numbers, short follow-up, and non-randomized designs. Nevertheless, they are biologically plausible given the association between type I interferon activation and pediatric NPSLE biomarkers. Future studies should include standardized neuropsychiatric attribution models, neurocognitive outcomes, CSF or blood interferon signatures, and neuroimaging endpoints.
Given the central role of type I interferon signaling in NPSLE pathogenesis, anifrolumab represents a promising candidate for targeted therapy; nevertheless, its precise role in CNS manifestations awaits confirmation in pediatric-specific studies.
B-cell directed therapies: Rituximab and beyond
B cells play a central role in the immunopathogenesis of neuropsychiatric systemic lupus erythematosus (NPSLE) through autoantibody production, antigen presentation, and cytokine secretion. Consequently, B-cell–targeted therapies have emerged as important options for patients with refractory disease, particularly when conventional immunosuppression fails.
Rituximab, a chimeric monoclonal antibody targeting CD20 on mature B cells, is widely used off-label in refractory pediatric and adult NPSLE. Multiple case series and retrospective cohort studies have reported favorable neurological outcomes following rituximab therapy, particularly in patients with seizures, psychosis, transverse myelitis, or cognitive impairment who are unresponsive to conventional immunosuppression. 74 Across these observational studies, approximately two-thirds of treated patients achieved clinical remission or substantial neurological improvement, often accompanied by reductions in disease activity scores and corticosteroid requirements. 75
The largest multicenter retrospective study to date evaluated 144 children with autoimmune and inflammatory central nervous system disorders, including a subgroup of patients with NPSLE. In this cohort, approximately 87% of patients demonstrated improvement in neurological outcomes as assessed by the modified Rankin Scale, encompassing motor, cognitive, and psychiatric domains. Rituximab was generally well tolerated, with infusion-related reactions and infections representing the most commonly reported adverse events. These findings support the potential efficacy of rituximab in pediatric autoimmune CNS disease, although disease-specific conclusions for NPSLE remain limited by subgroup size and retrospective study design. 76 However, this cohort included heterogeneous disorders, such as autoimmune encephalitis, demyelinating disease, vasculitis, and NPSLE. Because the number of NPSLE patients was limited and outcomes were not always reported separately for this subgroup, the overall response rate should not be interpreted as disease-specific efficacy for pediatric NPSLE. Rituximab may be more effective in antibody-mediated autoimmune encephalitis than in NPSLE phenotypes dominated by vasculopathy, thrombosis, or irreversible structural injury. Therefore, rituximab is best considered for selected inflammatory or B-cell–driven NPSLE phenotypes rather than for all neuropsychiatric manifestations.
Despite encouraging real-world data, randomized controlled trials in adults with systemic lupus erythematosus, most notably the EXPLORER trial, failed to demonstrate superiority of rituximab over standard therapy. 77 These discrepancies highlight the marked heterogeneity of NPSLE and underscore the importance of appropriate patient selection. Rituximab appears most effective in B-cell–driven inflammatory phenotypes rather than in vascular or non-inflammatory neuropsychiatric manifestations.
Obinutuzumab is a humanized type II anti-CD20 monoclonal antibody that induces more potent B-cell depletion than rituximab through enhanced antibody-dependent cellular cytotoxicity and direct cell death. Although its main evidence in SLE currently comes from lupus nephritis and systemic disease studies, a recent report has suggested its potential relevance as an alternative B-cell–depleting strategy in refractory SLE. 78 Its role in pediatric NPSLE remains undefined, and no dedicated pediatric NPSLE trials are available. Nevertheless, obinutuzumab may be considered a future candidate for patients with inadequate B-cell depletion, anti-rituximab antibodies, or rituximab intolerance, provided that safety, infection risk, hypogammaglobulinemia, and vaccine response are carefully monitored.
Other B-cell–directed and B-cell signaling strategies are also under active investigation. Preclinical studies suggest that BTK inhibition may attenuate neuroinflammation by disrupting pathogenic neuroimmune interactions, offering a promising therapeutic avenue for refractory NPSLE. Additionally, fully human anti-CD20 monoclonal antibodies, such as ofatumumab, may provide alternative options for patients who develop infusion reactions or anti-drug antibodies to rituximab, although clinical experience in pediatric NPSLE remains extremely limited. 79
In summary, CD20-directed B-cell depletion represents an important therapeutic option for selected patients with refractory pediatric NPSLE. While rituximab has demonstrated encouraging efficacy in observational studies, and obinutuzumab may represent a future alternative B-cell–depleting strategy, the absence of pediatric-specific randomized controlled trials and the heterogeneity of disease mechanisms necessitate cautious interpretation.
Chimeric antigen receptor T-cell (CAR-T) therapy in pediatric NPSLE
CAR-T therapy has recently emerged as a transformative approach for the treatment of refractory autoimmune diseases, including systemic lupus erythematosus. Early-phase clinical studies have demonstrated that CD19-directed CAR-T cell infusion can induce deep and durable remission in patients with severe, treatment-resistant SLE. In a phase I clinical trial reported in 2024, a dual-target BCMA–CD19 CAR-T construct achieved sustained, drug-free remission following a single infusion. 80 More recently, allogeneic CD19-targeted CAR-T therapy has also been shown to induce long-term remission and complete discontinuation of immunosuppressive therapy in refractory SLE. 81 A recent systematic review further supported the consistent efficacy and acceptable safety profile of CAR-T therapy in autoimmune diseases. 82
However, experience with CAR-T therapy in pediatric NPSLE remains extremely limited, and significant concerns persist regarding long-term safety, neurotoxicity, immune reconstitution, and feasibility. At present, CAR-T therapy should be considered experimental, with potential application restricted to highly selected, treatment-refractory cases within controlled clinical trial settings.
Emerging small-molecule and biologic therapies
Beyond cell-based approaches, several small molecules and novel biologics are under investigation to modulate pathogenic immune pathways in SLE.
Toll-like receptor 7/9 inhibitors
TLR7 and TLR9 play critical roles in nucleic acid sensing and type I interferon activation in SLE. Therapeutic inhibition of these pathways aims to suppress aberrant innate immune activation. IMO-8400, a synthetic oligonucleotide antagonist of TLR7/8/9, has demonstrated acceptable tolerability and preliminary efficacy in a phase two trial. 83 Although data specific to pediatric NPSLE are currently lacking, TLR pathway inhibition represents a rational strategy for interferon-driven disease subsets.
Dual BAFF/APRIL inhibitors: Telitacicept
B-cell survival factors BAFF and APRIL promote plasma cell maturation and autoantibody production. Telitacicept, a dual BAFF/APRIL inhibitor, represents a next-generation biologic designed to suppress both pathways simultaneously. Phase III trials and real-world studies have demonstrated significant efficacy and a favorable safety profile in patients with active SLE, leading to regulatory approval in China. 84 Furthermore, a real-world study reported that biomarker-guided telitacicept therapy achieved superior disease control in active SLE. 85 These results highlight telitacicept’s potential as a precision-oriented B-cell modulator.
Mesenchymal stem cell-based therapies
Mesenchymal stem cells (MSCs) exert immunomodulatory and tissue-repair effects through paracrine signaling and immune regulation. Clinical studies have reported short-term improvements in disease activity following MSC infusion in refractory SLE.86,87 Nevertheless, the durability of remission and the standardization of MSC preparation protocols remain major challenges for broader clinical translation.
JAK/STAT pathway inhibition
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway contributes to type I interferon-driven neuroinflammation in SLE. Therefore, tofacitinib, a JAK1/3 inhibitor, has been preliminarily observed to show efficacy in refractory childhood-onset SLE. In a retrospective case series involving nine pediatric patients with refractory cSLE, some individuals exhibited clinical improvement with acceptable tolerability and no severe adverse events. Nevertheless, dedicated trials for pediatric NPSLE are still lacking, and its therapeutic role in CNS manifestations remains investigational. 88
In summary, targeted therapies have substantially expanded the therapeutic landscape for pediatric NPSLE, offering opportunities to move beyond non-specific immunosuppression toward mechanism-based treatment strategies. While biologic agents targeting B cells and type I interferon pathways have shown encouraging results, robust pediatric-specific data—particularly for CNS involvement—remain limited. Emerging cellular and small-molecule therapies hold promise for refractory disease but require careful evaluation of long-term safety and efficacy. Together, these advances lay the foundation for precision medicine approaches tailored to the unique immunopathology of pediatric neuropsychiatric lupus.
Clinical challenges and strategies
Diagnostic challenges in pediatric NPSLE
The early diagnosis of pediatric NPSLE remains a substantial clinical challenge due to its heterogeneous manifestations and the lack of highly specific and validated biomarkers. 56 In children, NPSLE may present with seizures, cognitive impairment, mood disorders, psychosis, or even coma—clinical features that frequently overlap with infectious encephalitis, metabolic disturbances, and primary psychiatric disorders, thereby complicating timely recognition. 89 To date, no single laboratory test can reliably distinguish active inflammatory NPSLE from non-immune–mediated neuropsychiatric events. Conventional markers such as antinuclear antibodies (ANA), anti-double-stranded DNA (dsDNA) antibodies, and complement levels, are valuable for SLE monitoring but demonstrate limited sensitivity and specificity for CNS involvement. 90 Cerebrospinal fluid (CSF) analysis may reveal elevated protein levels or oligoclonal bands; however, these findings are nonspecific and occur in a wide range of inflammatory or infectious neurological conditions.
Neuroimaging further illustrates the diagnostic complexity. Conventional magnetic resonance imaging (MRI) frequently reveals white matter hyperintensities or basal ganglia abnormalities, yet similar radiographic changes may arise from hypertension, medication-related toxicity, or chronic ischemia. 91 Consequently, accurate diagnosis requires a multidisciplinary approach integrating clinical assessment, laboratory data, and neuroimaging findings. These limitations underscore an urgent need for advanced molecular and neuroimmune biomarkers capable of enabling earlier, more precise, and mechanism-informed diagnosis of pediatric NPSLE.
Limitations of current therapies
Current therapeutic strategies for pediatric NPSLE are constrained by several important limitations. First-line treatments typically involve high-dose corticosteroids combined with cyclophosphamide or mycophenolate mofetil as induction therapy, particularly in patients with severe systemic or central nervous system involvement. However, therapeutic responses vary widely, and a subset of patients continues to experience persistent or progressive neuropsychiatric symptoms despite standard regimens, necessitating escalation to alternative agents in refractory cases.89,92
Biologic therapies have expanded treatment options for refractory pediatric and childhood-onset SLE, but their application to pediatric NPSLE remains constrained by limited CNS-specific evidence. Most available data are derived from systemic SLE trials, adult NPSLE reports, retrospective cohorts, or mixed autoimmune CNS disease studies, and patients with active severe CNS involvement are often excluded from randomized trials.66,76 Therefore, treatment decisions must integrate neuropsychiatric attribution, inflammatory versus vascular phenotype, systemic disease activity, blood–brain barrier considerations, and individual safety risks.
Future research directions
Addressing the unmet needs in pediatric NPSLE requires coordinated efforts across several key research domains. First, the identification and validation of novel biomarkers is essential for precision diagnostics. Emerging evidence supports autoantibodies targeting neuronal antigens, including anti-NR2 glutamate receptor and anti-ribosomal P antibodies, cytokine profiles such as IL-6 and IFN-α, complement abnormalities, CSF inflammatory markers, and neuroimaging features as potential biomarkers for pediatric NPSLE.90,93 However, as highlighted in recent pediatric biomarker reviews, their diagnostic and prognostic performance remains insufficiently validated across independent pediatric cohorts. 90
Second, targeted therapies hold promise for enhanced efficacy and reduced toxicity. Anifrolumab, a type I interferon receptor antagonist, has demonstrated efficacy in adult SLE and is currently under evaluation in pediatric SLE through ongoing phase III trials, including the BLOSSOM study. Similarly, JAK inhibitors such as tofacitinib have shown preliminary benefits in small retrospective series of refractory childhood-onset SLE, suggesting potential applicability to interferon-driven CNS disease, although dedicated NPSLE trials are lacking. Finally, large-scale, multicenter, prospective cohort studies are critical to delineate the natural history of pediatric NPSLE and to refine evidence-based treatment algorithms. International consortia, such as the Paediatric Rheumatology International Trials Organisation (PRINTO), provide an important platform for harmonizing data across juvenile SLE cohorts. Integration of advanced neuroimaging techniques with multi-omics profiling may ultimately facilitate a transition from empirical management toward precision medicine in this complex condition. 56
Conclusion: A new era in the treatment of pediatric neuropsychiatric lupus
The management of pediatric NPSLE is entering a transformative phase, shifting from empirical, high-dose immunosuppression toward mechanism-driven and targeted therapeutic strategies. Over the past decade, deepened insights into immunopathogenesis—particularly the involvement of type I interferon signaling, complement activation, and neuronal autoimmunity—have paved the way for innovative biologic agents and small-molecule inhibitors. Belimumab, the first biologic approved for SLE in children aged ≥5 years, has demonstrated reductions in disease activity and corticosteroid exposure in pediatric SLE trials, although direct evidence in pediatric NPSLE remains limited. 66 Rituximab has shown encouraging effects in refractory autoimmune CNS disorders and selected NPSLE reports, but available data are largely observational and should not be generalized to all neuropsychiatric phenotypes. 76 More recently, the type I interferon receptor antagonist anifrolumab has shown efficacy in adult SLE and is currently under evaluation in pediatric phase III trials (e.g., the BLOSSOM study), warranting further investigation for NPSLE applications. JAK inhibitors, such as tofacitinib, have yielded preliminary benefits and acceptable safety profiles in small retrospective series of refractory childhood-onset SLE, highlighting a novel approach to modulating interferon-driven neuroinflammation. 88 In parallel, early reports of CD19-directed chimeric antigen receptor T-cell therapy suggest the potential for profound and durable remission in highly treatment-resistant SLE, although experience in pediatric NPSLE remains extremely limited. 94
Despite these advances, substantial evidence gaps remain. Pediatric-specific randomized controlled trials focusing on NPSLE are scarce, validated biomarkers predictive of treatment response are lacking, and the blood–brain barrier continues to limit central nervous system drug delivery. Addressing these challenges will require international multicenter collaboration, standardized outcome measures, and biomarker-guided clinical trial designs. This rapidly evolving landscape marks the dawn of a new era in precision medicine for pNPSLE, where the integration of multi-omics profiling, advanced neuroimaging, and AI-assisted models can potentially optimize therapeutic choices, minimize toxicity, and safeguard neurocognitive function in affected children.
Footnotes
Author contributions
Yunyan Li: writing – original draft (lead).
Shiling Zhong: writing – review and editing (equal).
Yuanling Chen: writing – review and editing (equal).
Ling Wu: writing – review and editing (equal),Funding acquisition(supporting)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Zhejiang Province Small but Strong Clinical Cultivation Innovation Team (Grant No.CXTD202502005)
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.