
Abstract
The observation of time to tumour progression (TTP) or progression-free survival (PFS) may be terminated by a terminal event. In this context, deaths may be due to tumour progression, and the time to the major failure event (death) may be correlated with the TTP. The usual assumption of independence between the TTP process and death, required by many commonly used statistical methods, can be violated. Furthermore, although the relationship between TTP and time to death is most relevant to the anti-cancer drug development or to evaluation of TTP as a surrogate endpoint, statistical models that try to describe the dependence structure between these two characteristics are not frequently used. We propose a joint frailty model for the analysis of two survival endpoints, TTP and time to death, or PFS and time to death, in the context of data clustering (e.g. at the centre or trial level). This approach allows us to simultaneously evaluate the prognostic effects of covariates on the two survival endpoints, while accounting both for the relationship between the outcomes and for data clustering. We show how a maximum penalized likelihood estimation can be applied to a nonparametric estimation of the continuous hazard functions in a general joint frailty model with right censoring and delayed entry. The model was motivated by a large meta-analysis of randomized trials for head and neck cancers (Meta-Analysis of Chemotherapy in Head and Neck Cancers), in which the efficacy of chemotherapy on TTP or PFS and overall survival was investigated, as adjunct to surgery or radiotherapy or both.
Keywords
1 Introduction
Dependent and informative censoring is a general issue that often arises in the analysis of censored data. And yet, most standard survival analyses are still based on the assumption of independence between time to event endpoints and the terminal event. When the independence assumption is questionable, the inference based on standard methodologies may be biased and possibly misleading. This dependence has been previously described in the analysis of progression-free survival (PFS) in oncology trials 1 and Food and Drug Adminstration's recent Draft Guidance for Industry on Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics recommends that patients who stop taking randomized therapy prior to documented progression are censored at the time when randomized treatment was stopped. 2 The rationale for this recommendation is not provided explicitly, but seems to be related to a concern that PFS times may be over-estimated or under-estimated otherwise. 3 A patient's death in the absence of documented progression remains an event, irrespective of whether the death occurred while the patient was still receiving randomized therapy or some time after stopping it. This approach ignores the issue of informative censoring. Patients who stop taking randomized therapy prior to documented progression frequently do so due to either toxicity of the drug or to a deterioration in the status of their disease. In such cases, the treating physician often judges that immediate intervention, commonly the introduction of a new cancer treatment, is in the best interest of the patient without necessarily waiting for confirmatory, radiographic evidence of progressive disease. PFS therefore can be censored informatively by death or dropout. Under those circumstances, if the prevalence of censoring differs between treatment arms, naive censoring could lead to extremely biased results and, ultimately, incorrect decisions. In total, there can be many reasons to use joint models of two survival endpoints, including to give a general description of the data, to correct for bias in survival analysis due to dependent dropout or censoring and to improve efficiency of survival analysis due to the use of auxiliary information. 4
Time to tumour progression (TTP) is defined as the time from randomization to the first event, between loco-regional progression or distant metastases, whereas PFS is generally defined as the time from randomization to the first event, between disease progression or death without documented progression, i.e. PFS = min(TTP,death). PFS is generally considered as a better surrogate for clinical benefit than other endpoints such as TTP alone, as the PFS endpoint includes overall mortality, so that unanticipated effects of treatment on survival are included in the endpoint. For the definitions of PFS as well as TTP, a careful and precise definition of tumour progression is crucial. 5 To assess whether PFS could be used as surrogate endpoints for overall survival (OS), research has focussed so far on empirical investigations of the correlation between estimates for PFS and OS.6,7 Censoring by death may be associated to worsening of symptoms, i.e. the patient is at higher risk of progression, while there could be situations where in fact censored patients are at less risk of disease progression, due for instance to a higher treatment benefit. Considering the definition of PFS (= the minimum of TTP and OS), it is clear that PFS and OS can hardly be fully independent since some information about OS is already contained in PFS. However, this dependency is less clear between TTP and OS, as deaths may be due to tumour progression. Although understanding the relationship between TTP and OS is highly relevant to the anti-cancer drug development, statistical models that try to describe the dependence structure between these two characteristics are not frequently used. We are aiming at filling this gap by considering a joint statistical model of TTP or PFS and OS. Fleischer et al. 8 have recently proposed a statistical model for the dependence between PFS and OS. However, their approach was completely parametrical and using separate hazard functions for the different times to events, in addition, the value of the correlation coefficient between PFS and OS itself was of limited (at best descriptive) value. This approach can only be a starting point for more sophisticated models that lead to a realistic description of the dependence structure between TTP/PFS and OS. Burzykowski et al. 9 proposed to use copulas models for the bivariate survival modelling. A progressive parametric multi-state model was also recently proposed by Dejardin et al. 10 assuming that progression always precedes death.
Meta-analytical approaches are clearly superior to single-trial approaches to study surrogate endpoints. Meta-analyses for the validation of surrogate endpoints have become a widely accepted and applied method in oncology research. 11 Without multi-trial data, it is almost impossible to make any direct inference about the association between the treatment effects and the surrogate and clinical endpoints, because one set of data cannot provide sufficient evidence of any association. Several authors have also indicated that a large sample size is required to evaluate potential surrogate endpoints. Multi-trial data readily have this capacity. In a large meta-analysis of randomized trials for head and neck cancers (Meta-Analysis of Chemotherapy in Head and Neck Cancers: MACH-NC), the efficacy of chemotherapy on OS was investigated, as adjunct to surgery or radiotherapy or both, the standard means to achieve loco-regional control.12,13 The Meta-analysis MACH-NC, based on individual patient data, compared loco-regional treatment with loco-regional treatment plus chemotherapy. The analysis showed a significant benefit on OS (HR = 0.88, [95% confidence interval (CI) 0.85–0.92]) of adding chemotherapy.
Association between PFS (or TTP) and OS depends on the disease and the characteristics of the population included such as tumour stage. For instance in old patients or in patients with major co-morbidities, deaths may be related to inter-current disease. In this case, death may often occur without a previous progression, i.e. a patient can die of inter-current disease while having a stable tumour or a tumour that responded to treatment.
In this article, we propose a general joint frailty model for clustered progression and terminal events. This approach is of interest for several reasons. First, it allows to deal with dependence between recurrent events and death for failure time data. It also gives information on whether TTP or PFS could be used as surrogate endpoints for OS. In addition, it allows a joint analysis of two processes which evolve with time, leading to more accurate estimates. This study extends previous research by dealing with clustered events (in meta-analytical studies) and giving smoothed estimates of the two hazard functions which represent incidence and mortality rates in epidemiology. It is natural in epidemiology to impose a continuous hazard function with small local variations. In this article, we propose a semi-parametric penalized likelihood method for estimating parameters in a joint frailty model for clustered and possibly recurrent data.
This article is organized as follows. In Section 2, we describe the joint frailty models. The construction of the full penalized log-likelihood is explained in Section 3. The model is applied to the joint analysis of TTP and OS in an individual patient data meta-analysis of chemotherapy in head and neck cancers (MACH-NC) in Section 4. Finally, section 5 presents a concluding discussion.
2 Joint model for times to progression and death in a meta-analysis
2.1 The models
To simplify the notation, we will set out only the joint models for one failure time and death; however, models can easily be proposed for the joint analysis of clustered recurrent failure times and death. Take the case of a study with G independent clusters (i = 1, … , G). We denote X
ij
as the survival time for subject j (j = 1, … , N
i
) from group i, C
ij
the corresponding censoring times (not by death) and D
ij
the corresponding death times. We first consider X
ij
as a time to event (or TTP). T
ij
= min(X
ij
, C
ij
, D
ij
) corresponds to each follow-up time and δ
ij
is a binary indicator which is 0 if the observation is censored or if the subject has died, and 1 if X
ij
is observed (δ
ij
= I(T
ij
=X
ij
), where I() denotes an indicator function). Similarly, we note
We assume continuous TTP, terminating, and censoring processes, so that progression and death cannot happen at the same time. We adopt the convention that death happens first in the small interval [t, t + dt). For subjects in the application study who died on the same day as their progression, they only count for terminal events, not as a progression.
Several formulations of the joint modelling of TTP and OS can be proposed:
– In the first setting, we assume that the association between TTP and OS is simply the result of individual associations or individual unmeasured factors. We consider the unobserved individual random effects ω
ij
(defined later). Following the previous model of Rondeau et al.,
4
the joint model for the hazard functions for TTP (r
ij
(.)) and death (λ
ij
(.)) for subject j from group i is thus:
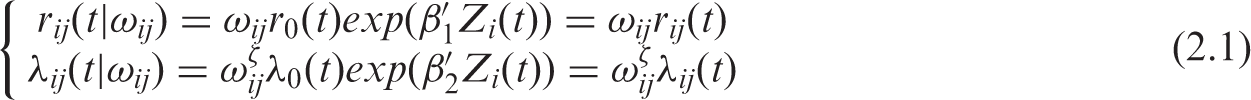
The random effects ω
ij
(frailties) are assumed independent. Mainly for reasons of mathematical convenience, the frailty terms are often assumed to follow a gamma distribution. The gamma frailty density is adopted here with unit mean and variance η. This choice and other possibilities such as log-normal, positive stable distributions are discussed in several papers.14,15 The dependence between
In the traditional model, the assumption is that ζ = 0 in (2.1), that is λ
ij
(t) does not depend on ω
ij
and thus death (or the terminal event process) is not informative for the failure rate r
ij
(t), i.e the two rates λ
ij
(t) and r
ij
(t) are not associated, conditional on covariates. When ζ = 1, the effect of the frailty is identical for the TTP and for the terminating event. When ζ > 0, the failure and the death rates are positively associated; higher frailty will result in a higher risk of progression and a higher risk of death. Inversely, when ζ < 0, higher frailty will result in a higher probability to develop a failure but in a lower survival. The interpretation of the value of ζ makes sense only when heterogeneity is present, i.e. when the variance of the random effects is significantly different from zero. However, in this model, we assume that the intra-cluster correlation is not present anymore after having taken into account prognosis factors and after adjustment for a subject-specific random effect term.
– In the second setting, we assume that the association between TTP and OS is the result of a clustered association (in our application, an intra-trial correlation). The second joint model is thus:
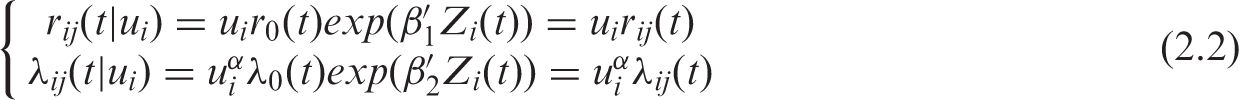
The random effects u
i
are assumed independent and gamma-distributed with unit mean and variance θ. The dependence between
The Kendall's tau can be used as a measure of dependency between the two outcomes of interest. Kendall's
6
τ
1
is the difference between the probability of concordance and the probability of discordance of two realizations of (
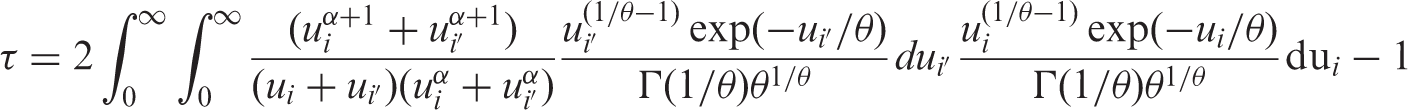
Model (2.2) can be generalized to:
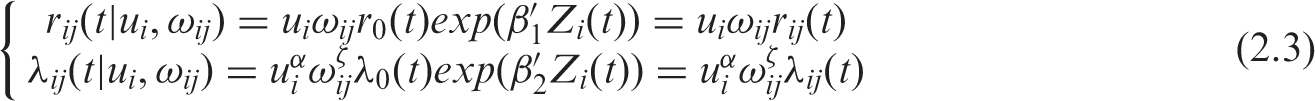
This generalization allows the baseline hazard functions used in (2.2) to be split into cluster- and subject-specific components. We assume here that the association between TTP and OS is both due to a clustered association and an individual association. We define here two random effects u i and ω ij and assume that the cluster-level random effects u i and the individual random effects ω ij are independent and gamma-distributed random effects Γ(1/θ; 1/θ) and Γ(1/η; 1/η) with E(u i ) = 1, var(u i ) = θ and E(w ij ) = 1, var(ω ij ) = η. The association or dependency between TTP and OS is then assumed to be the result of both individual- or cluster-level unobserved factors. These concepts are related to the individual- and trial-level surrogacy measures proposed in the framework of meta-analytic evaluation of surrogate endpoints. 17 We have not considered this generalization in the present application, due to the increased computational complexity of the problem.
2.2 Inference in the joint frailty model
We show the expression of the full log-likelihood for right-censored data for the model (2.2). The construction of the log-likelihood is detailed in Appendix 1. The expression can be easily extended to left truncated data (for instance if age is chosen as the basic timescale, see Appendix 2). In shared gamma-frailty models, the full log-likelihood for left-truncated and right-censored data takes a simple form with an analytical solution for the integrals.
18
This is not the case in the joint frailty model (2.2). The results should not be sensitive to the choice of frailty distribution, as supported for instance by Pickles and Crouchley's
15
simulation results. We denote φ = (r0(.), λ0(.), β, α, θ). To construct the likelihood function, apart from the usual assumption of independent censoring (other than death), the censoring must be non-informative for u
i
. We obtain the following expression of the full marginal log-likelihood in the joint gamma frailty model (2.2) for right-censored data:
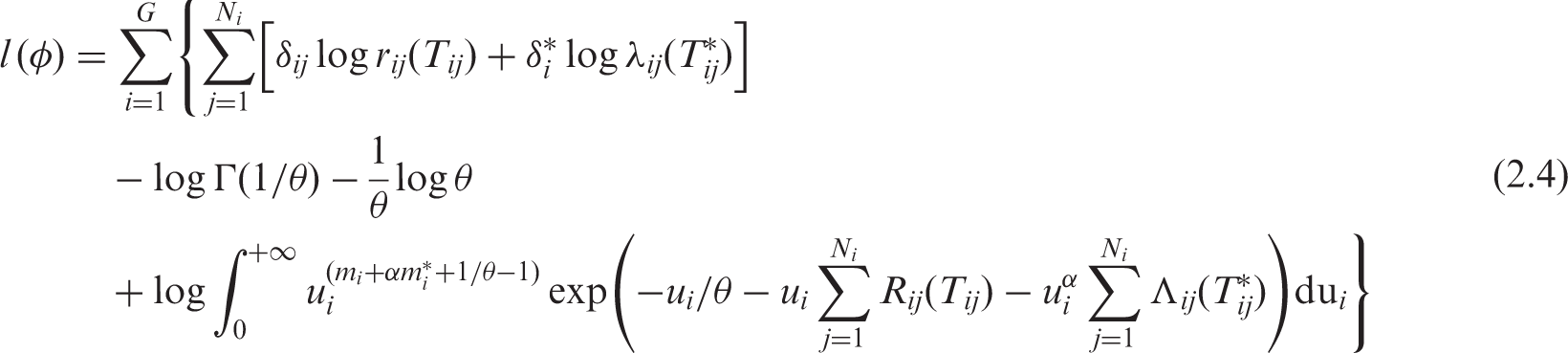
3 The semi-parametric penalized likelihood approach
We introduce a semi-parametric penalized likelihood approach to estimate the different parameters β, α, θ and the baseline hazard function r0(t) for TTP or λ0(t) for death times.4,19
In most situations, it is reasonable to expect smooth baseline hazard functions, piecewise constant modelling for the hazard functions being often unrealistic. To introduce such knowledge a priori, we penalize the likelihood by a term which has large values for rough functions.20,21 The roughness penalty function is represented by the sum of two squared norms of the second derivative of the hazard functions.
20
The penalized log-likelihood is thus defined as
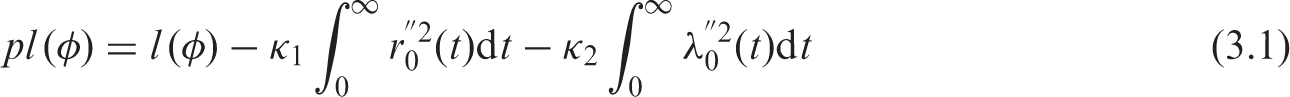
The estimators
We have previously shown that to obtain a good estimation of the theoretical hazard function, the more knots we used, the closer the MPnLE was to the true hazard function.
18
The smoothing parameters can be chosen by maximizing a likelihood cross-validation (LCV) criterion as in Joly et al.
21
Another approach consists in fixing the number of degrees of freedom to estimate the hazard function, as has been previously described.18,23 We thus use the relation linking the model degrees of freedom (mdf) and the smoothing parameter κ to evaluate the smoothing parameter:
We propose to maximize directly the observed log-likelihood (3.1) using a modified robust Marquardt optimization algorithm 24 which is a combination between a Newton–Raphson algorithm and the steepest descent algorithm. This algorithm is more stable than the Newton–Raphson algorithm 25 but preserves its fast convergence property near the maximum. The integrations in the full log-likelihood expression in (2.3) were evaluated using Gaussian quadrature. Laguerre polynomials with 20 points were used to treat the integration [0, ∞). To test whether the variance of the random effects was different from zero, i.e. H0 : θ = 0 versus. H a : θ > 0, the p-value from a Wald test was used. Because zero is the lower bound of θ, the unilateral Wald test was used, which is equivalent to a squared Wald test with a half-half mixture of zero and a chi-squared distribution with degree of freedom 1. 26
3.1 Software
The statistical software used was R and the library frailtypack (version 2.2-12) with the function frailtyPenal for the joint frailty models (2.1). 27 The frailtyPenal function was slightly modified to be able to handle joint modelling (2.2) and inserted in the R package ‘frailtypack’. Penalized likelihood maximization was used. In the reduced shared frailty models, κ1 and κ2 were evaluated using the cross-validation method, thereafter this value was used in the joint model. Furthermore, to deal with the constraint on the variance component (θ > 0 and η > 0), we used a squared transformation and standard errors of θ and η were computed by the Δ-method. 28 This reparametrization did not have any major adverse effect on the approximation, while being very numerically convenient. Since the initialization of the parameters is very important in this approach, we initialized the parameters with values obtained with simpler frailty models or Cox proportional hazard models.
4 Application to the meta-analysis MACH-NC
The meta-analysis MACH-NC, based on individual patient data, compared loco-regional treatment (as radiotherapy and/or surgery) with loco-regional treatment plus chemotherapy.12,13
We previously evaluated the heterogeneity between the underlying risks in trials and the heterogeneity of treatment effect between trials, using correlated random effects. 29 Results in the MACH-NC data confirmed a slight benefit for OS of adding chemotherapy to loco-regional treatment. A key issue when analysing these pooled data is to assess the dependency between TTP and OS. We thus aimed to investigate the proposed joint random effects models (2.1,2.2) for this updated MACH-NC data with treatment as a fixed effect (treated by chemotherapy versus not) and independent and gamma-distributed trial-specific or individual-specific random effects.
4.1 Data collection
Head and neck carcinomas (oral cavity, oropharynx, hypopharynx, nasopharynx and larynx) are frequent tumours for which surgery and/or radiotherapy are the main therapeutic agents. In the absence of a large (over 1000–2000 patients) randomized trial, the most reliable way to evaluate chemotherapy is to do a meta-analysis based on updated individual patient data. The initial Meta-Analysis of Chemotherapy on Head and Neck Cancer (MACH-NC) analysed data from nearly 11 000 patients in 63 randomized trials between 1965 and 1993. The first results showed an absolute benefit of 4% at 5 years in OS in favour of chemotherapy (p < 0.0001). 12 Most of the benefit was seen with concomitant radio-chemotherapy but with a relatively large heterogeneity in this subgroup of trials. The MACH-NC group decided to confirm the results by updating its database with the inclusion of the randomized trials performed between 1994 and 2000. Results were published in 2009. 13 Because some trials had strata that corresponded to different loco-regional treatments or chemotherapies, and because some trials had three-arms or a 2 × 2 design, a few trial arms were used twice, such that the number of comparisons in the meta-analysis was 108 and the number of patients was 17 493. The overall median follow-up was 5.6 years. The description of the dataset included and their references can be found in Ref. 13
As in Ref. 6, we pre-specified exclusion of two trials, which had no registered recurrences at all: one trial of induction chemotherapy (680 patients) and one trial of adjuvant chemotherapy (499 patients). We also excluded two very small trials (58 and 27 patients) of concomitant chemotherapy from the same institution, in which all patients were dead at 2 years. In MACH-NC, trials including only nasopharyngeal carcinomas were excluded.
4.2 Endpoints
Duration of TTP was defined as the time from randomization to the first event (loco-regional recurrence, loco-regional progression or distant metastases). Deceased patients were censored at the date of death. Patients without documented evidence of death were censored at the date of last follow-up. In all analyses, we used the locoregional or distant events as provided by the trial investigators, as defined by their own timing and assessment method. These definitions were heterogeneous across the included trials. For example, in some trials that investigated the addition of chemotherapy to radiotherapy as loco-regional treatment, patients who never reached complete remission after radiotherapy (defined by clinical or radiological assessment) were considered as having loco-regional failure at time zero, i.e. randomization.
PFS was defined as the time from randomization to the first event (i.e. locoregional, distant recurrence or death from any cause). Patients without documented evidence of an event were censored at the date of last follow-up. What we defined as PFS was often called disease-free survival in trials that included patients with resectable tumours and PFS in trials that included patients with non-resectable tumours. In the meta-analyses, both types of trials were present. There are also trials that included the mixed population. Therefore, we chose to apply our definition of PFS to all trials.
OS was defined as the time from randomization to death from any cause; patients still alive at the last follow-up visit were censored at the date of last follow-up.
During the data-collection process of the meta-analyses, central analyses of different types of events (i.e. loco-regional or distant relapses and deaths) were performed and sent out to the investigators for approval or modification.
4.3 Parameter estimates and interpretation
We analysed 16 099 patients from 103 treatment–control comparisons. The number of patients per trial varied between 25 and 573 with a median of 114 and a mean of 156. A total of 10 538 patients (65.3%) died and the number of deaths over trials ranged from 10 to 493 with a median of 74 and a mean of 102. A total of 8 314 patients (51.6%) experienced tumour progressions (between 5 and 487 by unit, with a median of 55 and a mean of 81).
Joint analysis of the TTP. PFS and death for the entire meta-analysis MACH-NC (1965–2000)

apl= Marginal penalized Log-Likelihood
bApproximate Cross-validation criterion (see Section 4.4)
Using simple shared frailty models (with cluster random effects) yielded a benefit of adding chemotherapy to loco-regional treatment for the TTP, the PFS and death. The variance of the cluster random effects were significantly different from zero for TTP, θ = 0.48 (SE = 0.07), for PFS, θ = .09 (SE = 0.02) and for OS, θ = 0.18 (SE = 0.03). When comparing joint model (2.2) to the reduced shared frailty models, the treatment effect on TTP is exactly the same, while the hazard ratio of chemotherapy on OS is closer to one in the joint model (2.2), but still significantly different from 1. Ignoring the dependence between time to the terminal event and TTP resulted in biases in the independent shared frailty model compared to the joint model. The same tendencies were observed for PFS. The intra-cluster association was lower in the shared frailty models compared to the joint model (2.2) (θ = 1.17 for TTP and θ = 1.10 for PFS). This can be explained by the fact that the variance θ of the cluster-specific random effects not only takes into account the intra-cluster correlation in the data but also the dependence between TTP and time to death.
In the joint frailty model (2.1), the chemotherapy treatment was also significantly associated with TTP and with OS, with the same tendencies. The chemotherapy treatment was no more significantly associated with PFS, nor with OS in the joint frailty model (2.1). The differences for TTP and OS between models (2.1) and (2.2) can be explained by their different modelling. Model (2.1) takes the individual over-dispersion into account, whereas model (2.2) takes into account the between trials heterogeneity.
In the joint frailty model (2.2), we observed that the treatment was more beneficial for TTP (RR = 0.88 (0.85–0.92)) than for OS (RR = 0.93 (0.89–0.97)). The treatment was also more beneficial for TTP than for PFS (RR = 0.93 (0.90–0.96)).
The joint models (2.1) and (2.2) showed significant variance of the random effects, meaning significant association between TTP and times to death, or between PFS and times to death, due to factors unobserved at the individual-level or at the cluster-level. We observe a positive association between the failure rates λ ij (t) and r ij (t) (measured by ζ > 0 or α > 0). This indicates that the incidence of progression is positively associated with death.
The values of Kendall's tau give moderate positive association between TTP and OS (or between PFS and OS) with τ = 0.37 (τ = 0.45) in the joint frailty model (2.1) and τ = 0.31 (τ = 0.33) in the joint frailty model (2.2) adjusted for the treatment effect. As expected by definition, the association between PFS and death is higher than the association between TTP and death.
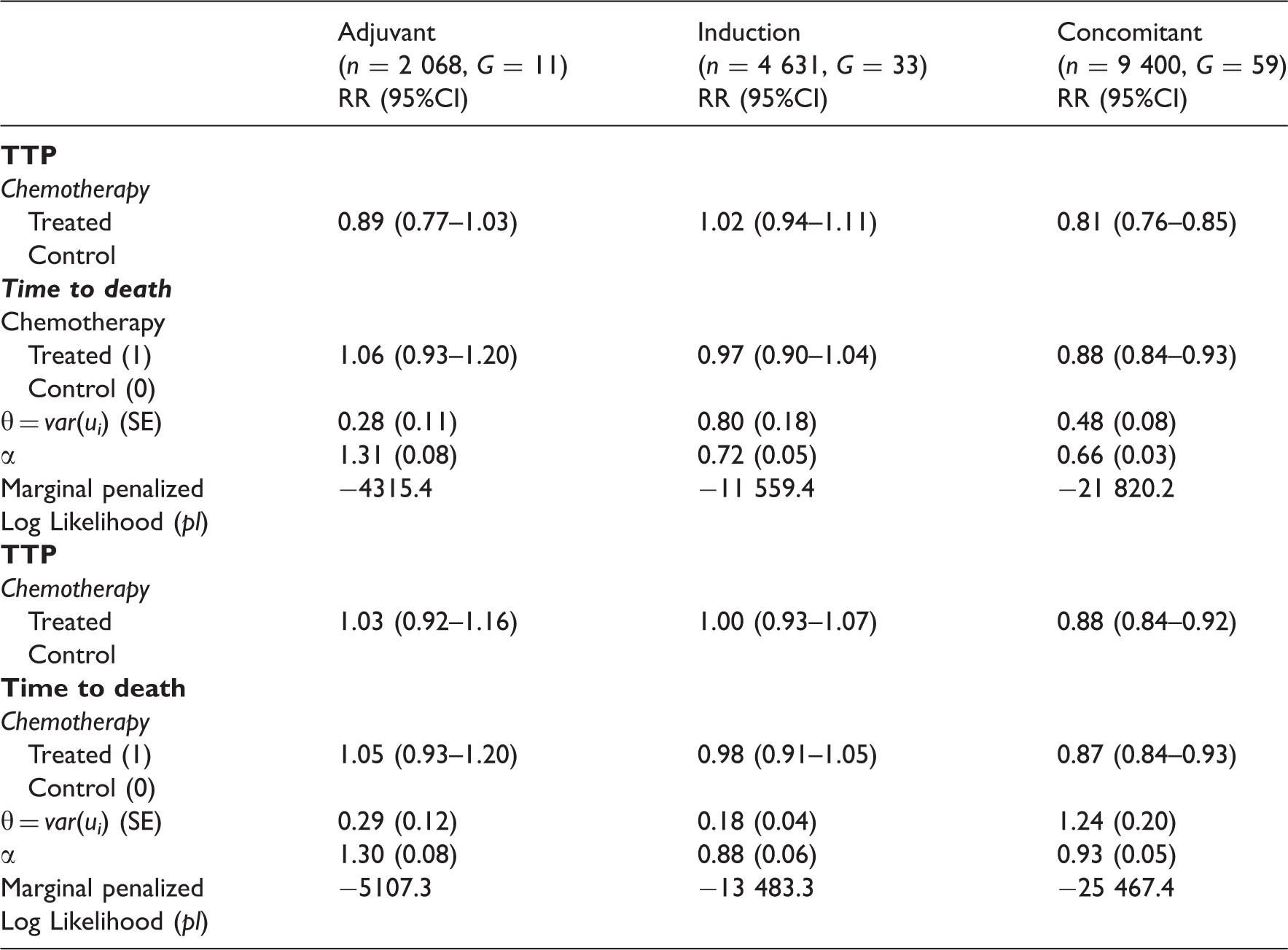
Joint analyses (using model 2.2) of the TTP and death or of the PFS and death for the treatment effect according to chemotherapy timing for the meta-analysis MACH-NC (1965–2000)
Joint analysis (using model 2.2) of the TTP and death with adjustment for different patient covariates for the meta-analysis MACH-NC (1965–2000)
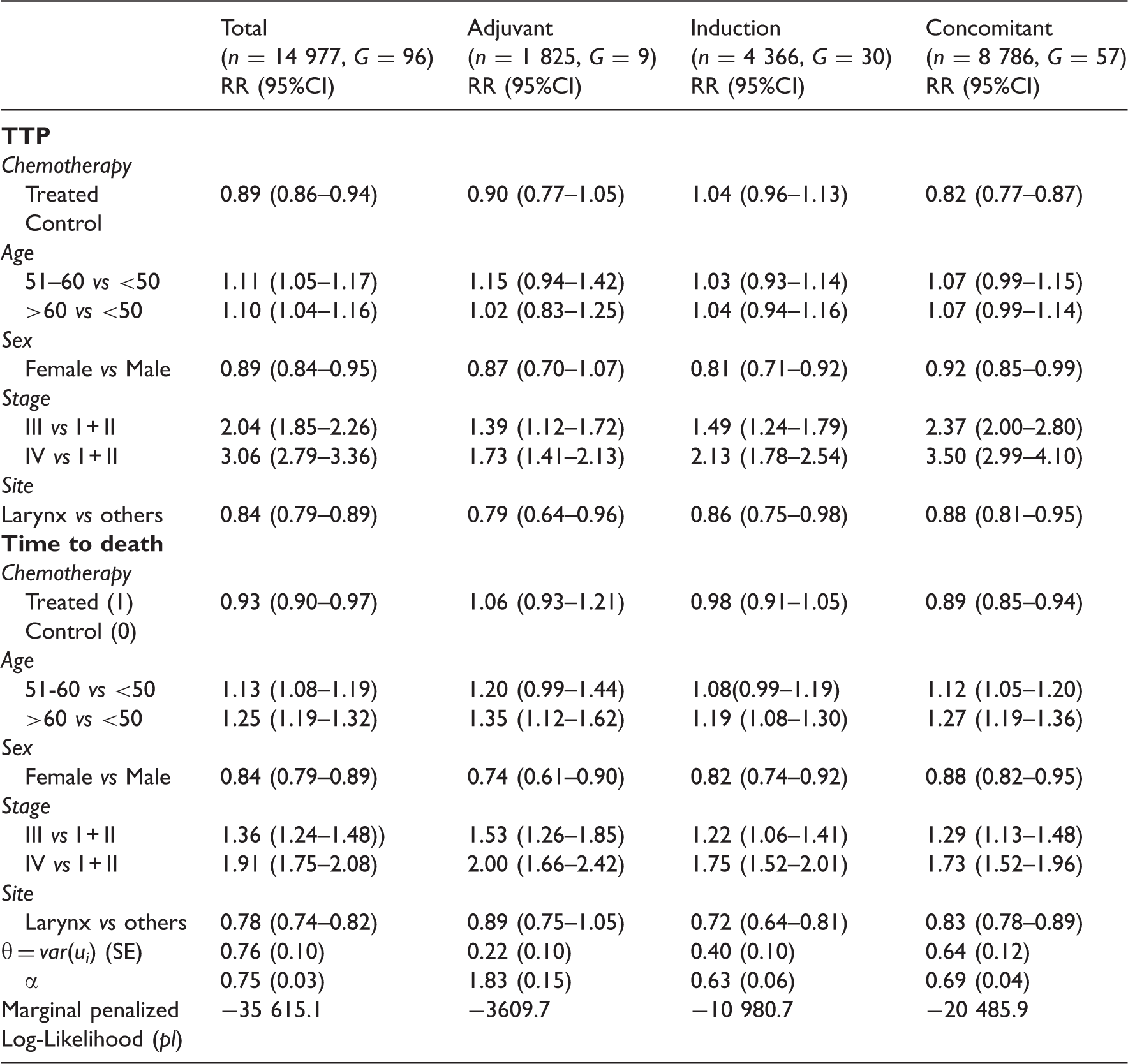
Figure 1 illustrates the survival functions for the three timing groups and according to the treatment. This confirms the greater survival benefit obtained with chemotherapy in the group of concomitant trials for the times to progression. We also observed a lower baseline survival for concomitant trials than for other timing of chemotherapy.
Survival functions for times to progression or time to death according to the chemotherapy and timing of the chemotherapy (in bold: treated) using joint frailty models (2.2).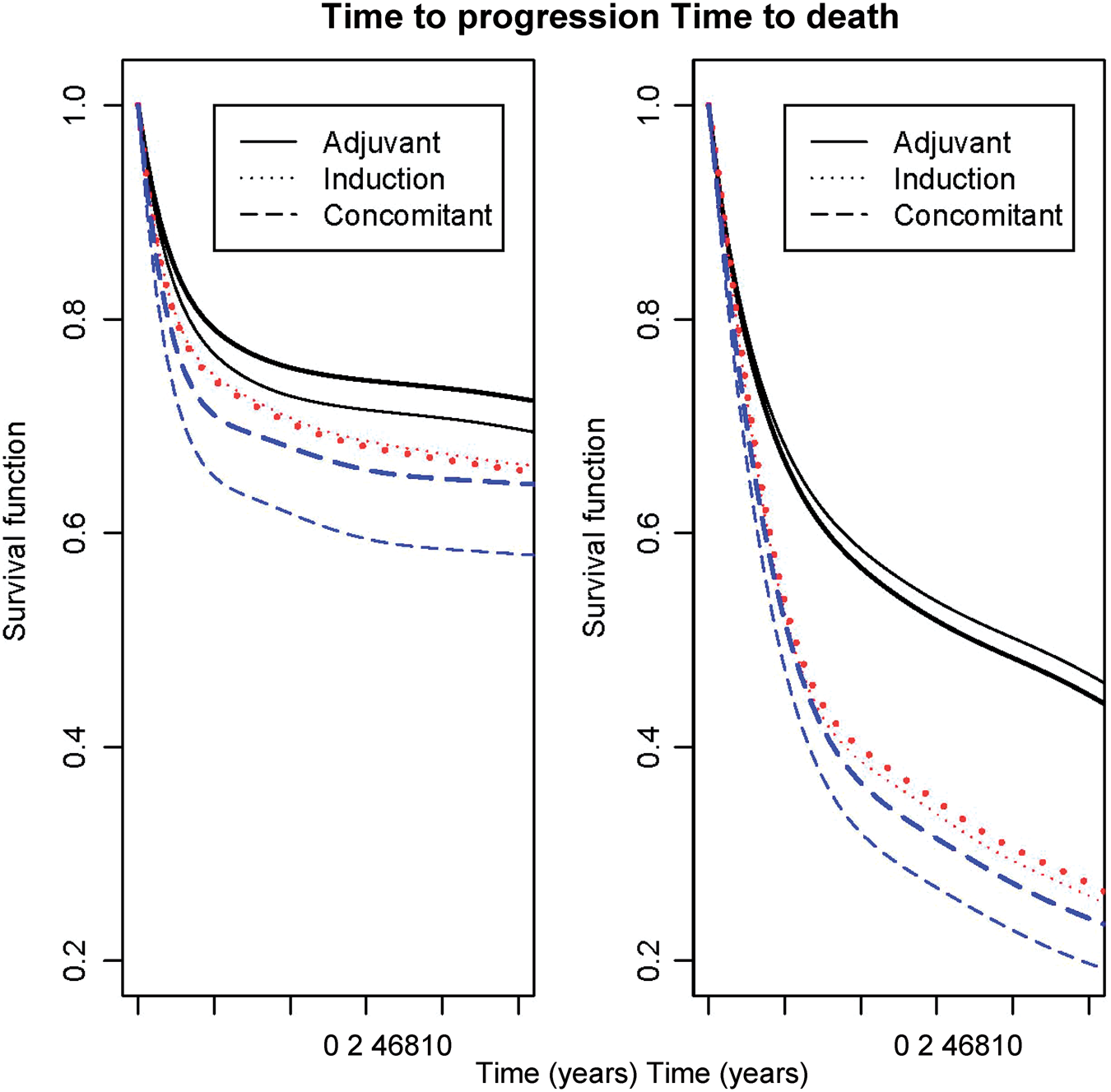
Even after adjusting for the patients' demographics (sex and age), the stage and the site of the initial tumour, there appeared to be a positive association between TTP and overall mortality due to unknown factors (Table 3). Adjustment for different individual covariates had no effect on the estimation of the overall treatment effect neither on its standard deviation. Very similar effects were observed for TTP and for OS in the joint models. When adjusting for covariates, the heterogeneity between trials decreased (from θ = 1.17 (0.15) to θ = 0.76 (0.10) in the joint model (2.2)) but remained significantly different from zero, this also indicates a significant association between the progression times and death.
We also performed a sensitivity analysis comparing recent trials, i.e. ending accrual between 1994 and 2000 (a more homogeneous group of trials mainly concomitant, with higher data quality and improved follow-up techniques) to the oldest (1965–1993). The observed percentages of progressions (45.8% vs 56.7%) or death (61.5% vs 70.3%) were lower in the more recent period than in the oldest. This is partly explained by the shorter follow-up for the more recent period, even if the follow-up techniques were improved. The magnitude of the benefit of the treatment was more important for the 1994–2000 trials (adjusted RR = 0.80 (0.74–0.86) for TTP and adjusted RR = 0.94 (0.88–1.00) for death) than for the 1965–1993 trials, especially for TTP (adjusted RR = 0.98 (0.91–1.05) for TTP and adjusted RR = 1.01 (0.95–1.08) for death). The difference between periods is explained by the change of proportion of concomitant trials over time. When the analysis is restricted to concomitant trials, the two RR values are similar. Some of the results presented in this article are different than those provided in the medical publications. 12 This is probably due to the fact that the previous simple survival models did not take into account the dependence with death, whereas the new joint survival modelling deals with this dependence and assumes that the association between TTP and OS is simply the result of unmeasured factors.
We also compared a penalized likelihood method of estimation with a parametric approach (i.e. using Weibull baseline hazard functions). The two methods gave similar estimations and standard deviations. The variance of the random effects (θ = 0.98 (0.12) in model (2.2) and η = 0.74 (0.01) in model (2.1)) and their corresponding coefficients (α = 1.05 and ζ = 3.27) were slightly under-estimated in the parametric approach compared to the semi-parametric approach (Table 1).
4.4 Model choice
A set of models were fitted in our analysis. For the random effects in the joint models, both trial- and individual-specific associations are considered and compared. Having included different types of random effects in the joint models for the two event-time outcomes, we obtained several candidate models from which a best-fitting model needed to be selected. The LCV criterion is adopted to guide the choice of the model used in this analysis. As LCV is particularly computationally demanding when n is large, an approximate version LCV
a
has been proposed by O'Sullivan
20
for the estimation of the hazard function in a survival case and adapted recently by Commenges et al.
30
Lower values of LCV
a
indicate a better fitting model. The LCV
a
is then defined as:
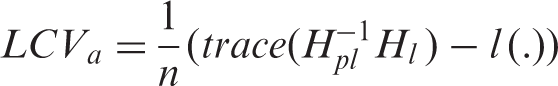
In the case of a parametric approach,
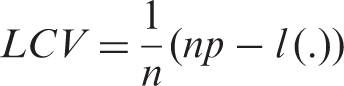
In Table 1, we report the values of LCV a for the joint models considered. The smallest LCV a , and therefore the best fit is achieved by the joint model (2.2) using a penalized likelihood estimation. This indicates that the joint association between TTP and times to death would be the result of unobserved factors at the trial level. Because the type of trial is probably correlated with both the ‘sickness’ of the patients and their survival times, linking the responses at the cluster-level would be more appropriate. This cluster-level link would indicate that trial could be a better surrogate for the unobserved process, and that a cluster-level link could be more likely to result in conditional independence of TTP and of times to death.
5 Discussion/Conclusion
In this article, we have developed a joint frailty model for clustered time-to-event data and a dependent terminal event. A penalized likelihood was used for the estimation of parameters in this joint model. Very similar effects were observed for TTP and for OS in the joint models, but with a higher beneficial effect of the treatment for TTP than for death. We observed a cluster- or trial-level link between the TTP and the times to death in the meta-analysis MACH-NC. As expected by definition, the association between PFS and death (measured with the Kendall's tau) is higher than the association between TTP and death. This link was more pronounced in the concomitant group of trials. This cluster-level link would indicate that trial is a better surrogate for the unobserved process.
The MACH-NC database might not be the most ideal database to answer the question of the impact of dependent terminal event in this specific clinical situation, but may be used for illustrative purposes. Indeed, the quality of the data is highly variable (some trials have a high proportion of patients who died of their cancer without date of progression before death). The trials were performed between 1965 and 2000 and the tools to detect progression have changed with time. After radiotherapy, it is sometimes difficult to know if a patient is in complete response or not, or to know when the loco-regional progression occurs. This problem was handled differently from one trial to the other. In some trials, the absence of complete response was considered as an initial loco-regional ‘progression’/failure, others used the change in tumour size or appearance of new nodes as loco-regional progression. At least, the quality of detection was the same in both arms.
In a meta-analysis combining survival data from different clinical trials, an important issue is the possible heterogeneity between trials. Such inter-trial variation cannot only be explained by the heterogeneity of the patients' baseline risk, but also by the heterogeneity of treatment effects across trials. Such a scenario can be accounted for using additive random effects in the Cox model, with a random trial effect and a random treatment by trial interaction, as we have previously proposed. 29 Thus, to include a random treatment by trial interaction in the joint model (2.2) would require complex correlation structure. While the model can be extended to other correlation structures, we chose not to do so, due to the increased computational complexity of the problem.
In this article, we have focussed on the cluster-level association between TTP (or PFS) and death in a meta-analysis of clinical trials using model (2.2). However, this association at the cluster-level may also be present in multicentre clinical trials. These two designs have differences: the meta-analysis will more often be international, with a larger sample size and with different protocols for each trial. In multicentre clinical trials, the participating centres may vary from university centres to community hospitals, so there is likely to be variation (heterogeneity) as well. The proposed model and estimation approach are of particular interest for both designs in order to study the heterogeneity when the sample size is sufficient.
In analyses of the natural history of cancer, there is a great interest in dynamic prediction of death, that is, in the computation of the predictive distribution of death at a certain moment in time given, the history of event(s) (ex: local or distant relapse) and covariates until that moment.31,32 The interest is then to produce an accurate estimate of the probability to survive beyond t + Δt, conditional on information available at the prediction time point t. The joint models described here could be an interesting prognostic tool to dynamically assess a patient's prognosis of death using recurrence information. To more accurately guide clinical decision making, the monitoring of recurrences after initial treatment would be facilitated by dynamic powerful prognostic tools that incorporate the complete post-treatment recurrence evolution. It is worth to note that any investigations on the dependency structure between OS and TTP can only be reasonable if the measurement of TTP itself is accurate. Therefore, before thinking about predictions for OS, issues like the determination of progression and interval censoring of TTP need to be taken into account. Furthermore, in our application, we censored patients at their date of last follow-up or at their date of death. We then assumed that patients were actively followed up for their progression until their date of censoring. This assumption can be accurate for some trials but more questionable for other trials.
Another application of these joint frailty models would be to study the specific causes of death, i.e. cancer-death instead of OS. However, from a modelling point of view, this would require the modelling of three functions instead of two: one for TTP, one for deaths related to head and neck cancers and one for non-cancer deaths. This extension could be a little bit computationally challenging. Recent publication on the same meta-analysis has shown that the benefit of chemotherapy was mostly due to its effect on deaths related to head and neck cancers (HR = 0.78 [0.73–0.84], p < 0.0001) and had no effect on non-cancer deaths (HR = 0.96 [0.82–1.12], p = 0.62). 13
The likelihood cross-validation criterion (LCV a ) measures only the relative goodness of fit among a collection of models. Although LCV a contributes to the relative identification of the best-fitting model, it provides no information about the absolute adequacy of the models. Thus, other model diagnostic tools are needed to assess the adequacy of the model. A future research topic would be to study model checking, using for instance martingale residuals.
Footnotes
Acknowledgements
The authors thank the trialists and research group who agreed to share and update their data and the following institutions for funding the investigators' meetings or the meta-analysis project: Association pour la Recherche sur le Cancer, Programme Hospitalier de Recherche Clinique, Ligue Nationale Contre le Cancer, Sanofi-Aventis (unrestricted grants), University College London Hospitals/University College London Comprehensive Biomedical Research Centre.
Trialists and research group
D.J. Adelstein (Cleveland Clinic Foundation), J.P. Armand (Institut Claudius Regaud), C. Amand (Intitut Gustave-Roussy [IGR]), H. Audry (IGR), J.M. Bachaud (Institut Claudius Regaud), H.G. Bartelink (Netherlands Cancer Institute, Amsterdam), J. Bernier (Clinique de Genolier), W.R. Bezwoda (University of the Witwatersrand), A. Buffoli (Institituto di Radioterapie Oncologica, Udine), J. Bourhis (IGR), K.T. Bhowmik (Vardhaman Mahavir Medical College), D. Brizel (Duke University Medical Center), G.P. Browman (Mc Master University), V. Budach (Charité University Medicine Berlin), G. Calais (Centre Hospitalier Universitaire de Tours), B.H. Campbell (Medical College of Wisconsin), A. Carugati (Instituto de Oncología Ángel H Roffo), S. Chalkidou (Hellenic Cooperative Oncology Group), J.R. Clark (Dana Farber Cancer Institute), E. Cohen (Institut Claudius Regaud), L. Collette (European Organization for Research and Treatment of Cancer), O. Dal Canton (Ospedale S Giovanni Vecchio, Torino), D. Dalley (St. Vincent's Hospital, Sydney), J. Depondt (Centre Hospitalier Universitaire de Bichat), L. Désigné (IGR), A. Deszcz-Thomas (Centre Hospitalier Universitaire de la Pitié-Salpêtrière), B. Di Blasio (Parma University Hospital), W. Dobrowsky (University of Vienna), C. Domenge (IGR), F. Eschwege (IGR), J.F. Evensen (Norwegian Radium Hospital), Radiation Therapy Oncology and Head and Neck Cancer Groups of the European Organisation for Research and Treatment of Cancer, C. Fallai (Università di Firenze), J.J. Fischer (Yale University), A.A. Forastiére (Johns Hopkins University), G. Fountzilas (Aristotle University of Thessaloniki), K.K. Fu (University of California San Francisco), D. Gedouin (Centre Eugène Marquis), S. Guérin (IGR), F. Geara (MD Anderson Cancer Centre), R. Giglio (Instituto de Oncología Ángel H Roffo), N.K. Gupta (Christie Hospital, Manchester), E. Haddad (Hôpital Universitaire H Mondor), B.G. Haffty (UMDNJ-Robert Wood Johnson Medical School), Y. Hasegawa (Aichi Cancer Centre), C. Hill (IGR), J.C. Horiot (Centre Georges François Leclerc), M. Horiuchi (Tokaï University), J. Houghton (Cancer Research Campaign & University College London Cancer Trials Centre), P. Huguenin(Deceased) (University Hospital Zurich), P. Jan (IGR), Ch. Jaulerry (Institut Curie), B. Jeremic (Kragulevac University Hospital), T. Jouffroy (Institut Curie), A. Jortay (Brumann University Hospital), B. Kapstad (University of Bergen), L.P. Kowalski (A.C. Camargo Hospital, Sao Paulo), S. Kramer (Thomas Jefferson University Hospital), K.S. Krishnamurthi (Madras Cancer Institute), S. Kumar (Sanjay Gandhi Post Graduate Institute of Medical Science), G. Laramore (University of Washington, Seattle), E. Lartigau (Centre Oscar Lambret), J.L. Lefebvre (Centre Oscar Lambret), A. Le Maitre (IGR), T. Leong (Harvard University), F. Lewin (Huddinge University Hospital), B. Luboinski (IGR), M. Luboinski (IGR), E. Maillard (IGR), T. Maipang (Prince of Songkla University), M. Martin (Centre Hospitalier Intercommunal de Créteil), J.J. Mazeron (Centre Hospitalier Universitaire de la Pitié-Salpêtrière), M. Merlano (National Institute for Cancer Research, Genoa), S. Mehta (Mumbai Group), R. Molinari (Istituto Nazionale Tumori, Milano), K. Monson (Cancer Research Campaign & University College London Cancer Trials Centre), K. Morita (Aichi Cancer centre), R.P. Mueller (University of Wuerzburg), A. Murias (Hospital Insular del Gran Canaria, Las Plamas), V. Mosseri (Institut Curie), G. Numico (National Institute for Cancer Research, Genoa), P. Olmi (Università di Firenze), B. O'Sullivan (Princess Margaret Hospital, Toronto University) J. Overgaard (Aarhus University Hospital), A. Paccagnella (SS Giovanni and Paolo Hospital, Venezia), T. Pajak (Radiation Therapy Oncology Group), L.M. Parvinen (Turku University Hospital), N.W. Pearlman (Denver VA Medical Center), J.P. Pignon (IGR), R.S. Rao (Tata Memorial Hospital, Bombay), J.M. Richard (IGR), K. Rufibach (Swiss Group for Clinical Cancer Research), F. Sanchiz (I Policlinico, Barcelona), H. Sancho-Garnier (Centre Val d'Aurelle), D.E. Schuller (Ohio State University Hospital, Columbus) V. Shanta (Madras Cancer Institute), R.J. Simes (NHMRC Clinical Trials Center, Camperdown), L. Smid (University of Ljubljana), L.A. Stewart (University of York), S. Staar (University of Cologne), P. Strojan (Institute of Oncology Ljubljana), H. Stuetzer (University of Cologne), H. Szpirglas (Centre Hospitalier Universitaire de la Pitié-Salpêtrière), G. Schwaab (IGR), N. Syz (IGR), S. Takaku (Saitama Medical School), S.G. Taylor (Rush Medical Center), J.S. Tobias (Cancer Research Campaign & University College London Cancer Trials Centre), R.J. Toohill (Medical College of Wisconsin), E.E. Vokes (University of Chicago Medical Center), P. Volling (University of Cologne), M.C. Weissler (University of North Carolina, Chapel Hill), T. Wendt (University of Jena), K.D. Wernecke (Charité University Hospital), J. Widder (University of Vienna), G.T. Wolf (University of Michigan) and K. Yoshino (Osaka Medical Center for Cancer and Cardiovascular Disease).