
Abstract
Early phase trials of complex interventions currently focus on assessing the feasibility of a large randomised control trial and on conducting pilot work. Assessing the efficacy of the proposed intervention is generally discouraged, due to concerns of underpowered hypothesis testing. In contrast, early assessment of efficacy is common for drug therapies, where phase II trials are often used as a screening mechanism to identify promising treatments. In this paper, we outline the challenges encountered in extending ideas developed in the phase II drug trial literature to the complex intervention setting. The prevalence of multiple endpoints and clustering of outcome data are identified as important considerations, having implications for timely and robust determination of optimal trial design parameters. The potential for Bayesian methods to help to identify robust trial designs and optimal decision rules is also explored.
1 Introduction
Complex interventions contain several distinct and potentially interacting components, each of which may contribute to the efficacy of an intervention as a whole.
1
For example, psychotherapy may be viewed as being composed of two treatment variables, namely techniques described in a therapy manual together with a therapist delivering these techniques.
2
This contrasts with typical drug treatments, where the drug is the only treatment variable to consider. While drug regimens may be complex,
3
randomisation and blinding allow the effects of a drug to be separated from the context in which it is provided. Broad classes of complex interventions include surgical, behavioural, psychological, educational and physical interventions. The evaluation of a complex intervention raises specific challenges, and several frameworks have therefore been proposed to guide this process. These include a widely used framework proposed by the MRC1,4 the IDEAL initiative aimed at surgical interventions;
5
and the Multiphase Optimisation STrategy (MOST).6,7 The most recent MRC framework is summarised in Figure 1.
Current MRC guidance on the development and evaluation of complex interventions, adapted from Craig et al.
1
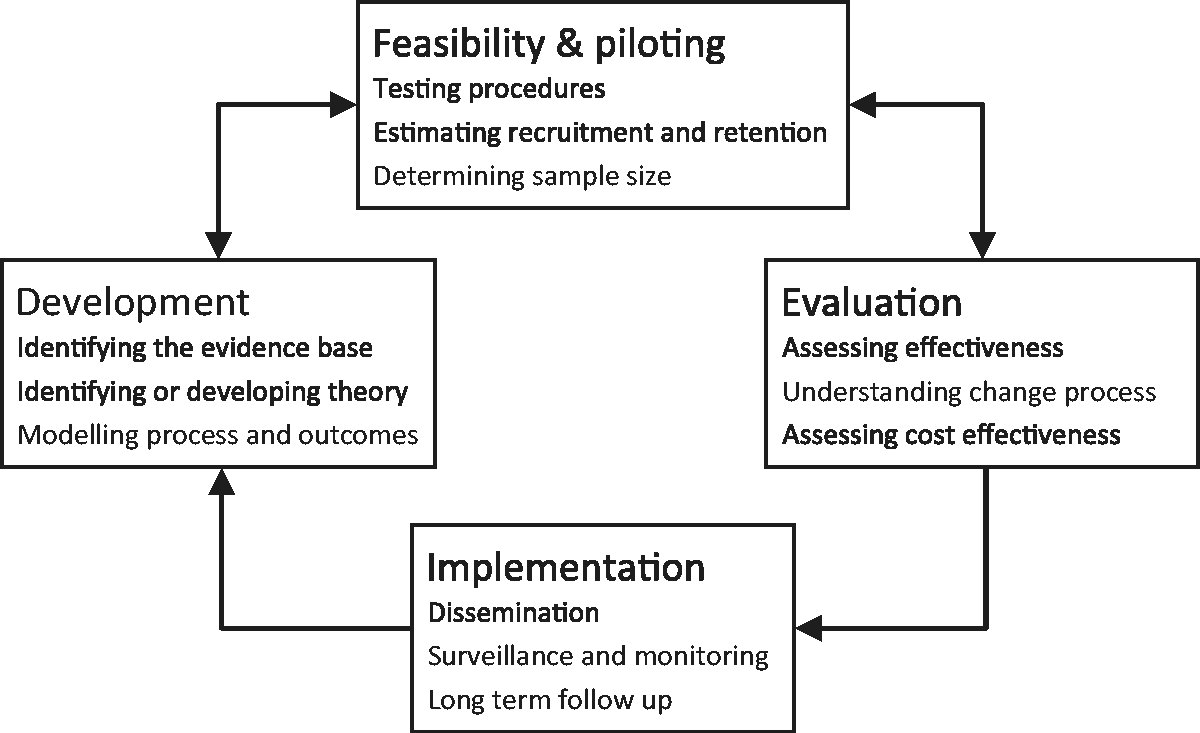
As shown in Figure 1, ‘feasibility and piloting’ is identified as one of four key stages in the development and evaluation of complex interventions. While the definitions of, and distinctions between, feasibility and pilot studies are not always clear,8–10 the MRC guidance states that the purpose of this stage is to inform the design of a subsequent large, definitive trial assessing the effectiveness of the intervention. Several parameters required for designing the definitive trial may be estimated at this stage, including the variance of the proposed outcome measure(s), recruitment and follow-up rates, and intra-class correlation coefficients (ICCs) in trials with clustering effects. Characteristics relating more directly to the intervention, such as its acceptability and the level of adherence, may also be assessed. Gathering information relating to these factors reduces the likelihood of the large trial failing due to poor design.
While MRC guidance recommends evaluating a complex intervention following feasibility or pilot work, in practice it is not uncommon for feasibility or pilot studies to include evaluation through hypothesis testing. For example, a recent review found that 21 of 26 feasibility and pilot studies surveyed included a hypothesis test. 8 However, the size of these studies is often derived using generic rules of thumb11,12 rather than through formal power calculations, with the review finding that only 9 of the 26 studies reported a sample size calculation. 8 As a result, hypothesis tests are likely to be underpowered 13 and the typical recommendation is that such tests should be de-emphasised, interpreted with extreme caution, or avoided altogether.8,13–15
It could be argued, however, that a formal assessment of potential efficacy or activity should be carried out in pilot and feasibility studies, and that such studies should be properly designed to address this objective. In this manner, feasibility or pilot work could not only ensure that subsequent large scale randomised controlled trials (RCTs) of complex interventions are well designed, but could also reduce the rate at which such trials fail due to an inherently ineffective intervention. Moreover, this approach would clearly be more efficient than conducting a feasibility or pilot study and then a separate study assessing only efficacy. To begin developing such designs, one may look to methods developed in the drug setting. There, small ‘phase II’ trials which focus on making an early assessment of efficacy, identifying promising and discarding unpromising drug treatments, are commonplace.
The application of phase II designs to the complex intervention setting is not straightforward due to challenges that are commonly encountered in complex intervention trials. For example, the assumption that patient outcomes are statistically independent is often violated as a consequence of cluster randomisation, 16 a group-based intervention 17 or therapist variation. 18 The associated implications for precision are compounded in cases where only a small number of clusters are available, as is often the case in feasibility or pilot studies. 19 Furthermore, the multi-component nature of complex interventions will mean that an assessment of efficacy will often have to be based on multiple endpoints, 1 in contrast to the single binary indicator of ‘success’ often used in phase II studies.
An example which serves to illustrate each of these points is the OK-Diabetes (OK-D) feasibility trial of a supported self-management intervention for adults with type II diabetes and learning disabilities. 20 The OK-D study involves first developing a manualised intervention and then carrying out a randomised feasibility study whose objectives include estimation of recruitment rates, testing of data collection forms and assessment of the feasibility of delivering the intervention. While the feasibility study individually randomises treatment packages to patients, diabetes specialist nurses provide the intervention and may, therefore, induce a clustering effect in the intervention arm. Furthermore, as the intervention is newly developed only two nurses will be involved. The intervention is targeted at three aspects of poor diabetes self-management, and as a result there are a number of possible outcomes to be considered when assessing efficacy.
It has been proposed that the OK-D feasibility trial be extended to allow for a preliminary assessment of the efficacy of the developed intervention as a formal objective, highlighting the need for appropriate trial design methodology. In this paper, we will review the approach to assessing efficacy developed in the context of phase II trials for drug therapies, setting out the key methodological challenges to their application in feasibility and pilot studies of complex interventions, and thereby outlining future directions for methodological research. In section 2, an overview of phase II designs and their key characteristics will be provided. In section 3, multiple endpoints and clustering will be discussed in detail, considering the formulation of decision rules, difficulties arising from nuisance parameters, and practical difficulties in determining sample size in a timely manner. Finally, in section 4 conclusions are drawn and further avenues for future research are suggested.
2 Efficacy evaluation in oncology drug trials
Following the determination of a safe dose in phase I, but before a definitive RCT in phase III, phase II trials typically act as a screening mechanism to screen out ineffective drugs at an early stage and progress only the most promising treatments to phase III. Phase II designs tend to employ a decision-focussed approach to inference, with an emphasis on determining if a subsequent phase III trial is warranted as opposed to estimation of underlying parameters. This approach is typically sustained through the use of Neyman–Pearson hypothesis testing or, alternatively, through Bayesian decision-theoretic methods. 21
In the case of hypothesis testing, trial design focusses on ensuring type I and II error rates remain within pre-specified nominal bounds. Perhaps the simplest phase II design to employ hypothesis testing for a single binary outcome was proposed by Fleming,
22
then extended by A’Hern
23
from an approximate to an exact test. To use the design, one must first specify a success rate p0 which, if true, would mean the new intervention would not be worthy of further investigation. An alternative hypothesis pA must then be given, corresponding to a success rate which would certainly merit a full evaluation in a definitive RCT. Applying this to the OK-D problem, we could set
A wide range of alternative phase II designs have been published, accounting for the variety of problems to which they may be applied. 24 Only a brief overview of the main differences between designs, with references to examples, is considered here. One point of differentiation between the designs is in the number of stages. While the A’Hern 23 design described above involved a single decision point, designs such as those proposed by Simon 25 include an additional interim analysis to allow for the phase II trial to terminate early due to futility. Single-arm designs may be contrasted with randomised designs, 26 which allow a concurrent as opposed to historical control to be used. Multi-arm designs, for cases where multiple treatments are available for evaluation at once, have also been described. 27 While the majority of phase II designs focus on a single endpoint relating to efficacy, several have been proposed which can consider additional measures relating to, for example, toxicity28,29 or further aspects of efficacy. 30
In addition to hypothesis testing designs, there are also a number which adopt a Bayesian framework. These vary in the extent to which Bayesian methodology is employed, from allowing some prior information to be incorporated in the form of probability distributions 31 to full decision-theoretic frameworks.32,33 The multitude of designs available requires a thorough assessment of the key design criteria specific to the trial in question, to ensure an appropriate design is selected.
3 Efficacy evaluation in complex intervention trials
When applying ideas from phase II trials in an early phase complex intervention setting, it is important to take account of complexities relating to (i) prevalence of multiple endpoints and (ii) recruitment- and treatment-related clustering effects.
3.1 Multiple endpoints
Multiple endpoints, on which the decision of proceeding to phase III should be based, arise due to several reasons. In addition to an assessment of efficacy requiring several endpoints, due to the multi-component nature of the intervention, endpoints relating to safety, acceptability and adherence are often required. Further to these, endpoints relating to the feasibility of a phase III trial, such as measurements of the recruitment and follow-up rates, should be taken into account. Thus, while phase II drug trials are not always limited to a single endpoint, early phase evaluations of complex interventions may routinely involve more.
As an example, the original design of the OK-D feasibility study included three feasibility criteria which were to be met to consider progression to phase III. These took the form of threshold values of recruitment rate, numbers lost to follow up, and adherence of participants in the intervention arm. In addition to these three endpoints, a further four endpoints were of interest in terms of assessing efficacy. Specifically, continuous measurements of glycated haemoglobin (HbA1c), blood pressure, total cholesterol and body mass (BMI) were all proposed as potential efficacy endpoints, with no single one anticipated to be sensitive to all components of the intervention.
3.1.1 Decision rules
In the single-endpoint case, a decision rule regarding progression to a phase III trial can be defined by a single cut-off point, as was illustrated in the example in section 2. Where several endpoints are present, specifying the form of the decision rule for progression to phase III becomes more complex. This problem has been addressed to a limited extent in the drug setting. In the case of two binary endpoints describing efficacy and toxicity, phase II designs such as that of Bryant and Day
28
consider separate null and alternative hypotheses for the two endpoints, resulting in four ‘states of nature’. Specifically, defining ‘unacceptable’ and ‘acceptable’ levels of both efficacy and toxicity as Example decision rules arising from phase II designs for two endpoints, where the shaded area of the sample space represents the decision to progress to phase III: (a) 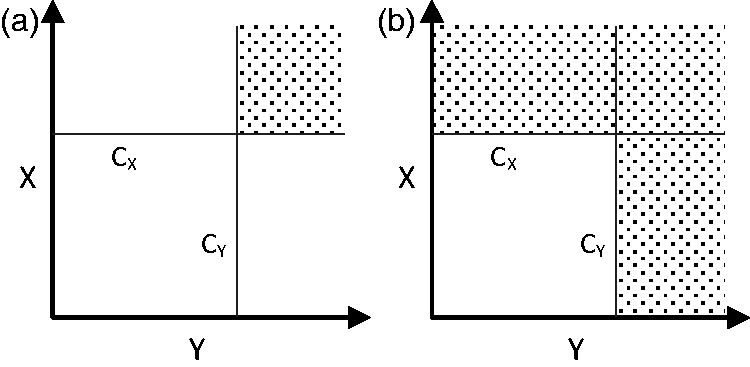
In cases where two binary measures of efficacy are of interest, phase II designs such as that of Sill et al. 30 employ a rule whereby we proceed to phase III if either quantity reaches the specified cut-off point. The form of the resulting acceptance region is illustrated in Figure 2(b). Again, the form of this rule dictates the possible types of errors, with a single type I error rate in this case and two type II error rates. One advantage of these decision rules is the ability to discriminate between endpoints through their nominal error rates. For example, in the case of the Bryant and Day design, progressing to phase III with a toxic treatment may be considered more of a risk than progressing with an ineffective treatment, and so the nominal type I error rate relating to toxicity could be set to a lower level to ensure this error is less likely. Similarly, in the case of two efficacy endpoints and the Sill et al. design, one could set the nominal type II error of the preferred endpoint to be lower than the other to ensure the trial will be more likely to detect a treatment which is promising in this respect.
Beyond this use of nominal error rates, designs such as those of Bryant and Day
28
and Sill et al.
30
do not provide any means with which to describe any relative preferences between different qualities of the treatment. With multiple endpoints to consider, it is possible that the decision to progress to phase III could involve trading off one aspect of the treatment against another. For example, one may be happy to accept a slightly toxic treatment if it demonstrated substantial efficacy, but not if efficacy was only moderate. Phase II designs allowing for such a trade-off were proposed by Conaway and Petroni.34,35 Considering binary efficacy and toxicity endpoints with parameters pE and pT, the authors propose dividing the parameter space
An alternative approach to acknowledging the presence of multiple endpoints is proposed by Sargent et al.
38
In this phase II design, the decision space related to the trial is expanded from {stop, go} to include a third, intermediate decision. Considering an explicit primary endpoint, if the corresponding observations are strong enough (in either direction) the trial will lead to one of stop or go. If the observations are less conclusive, it is suggested that the decision should now be made by considering other endpoints of interest. This design therefore provides a formal mechanism to allow for the inclusion of more than one endpoint without requiring any specification of their nature or relationship to one another at the design stage. All that is assumed is that a partial ordering of preference exists, with the primary endpoint considered more important than all other endpoints. As such, it represents a flexible methodology which could be applied to the complex intervention setting where many endpoints are of interest. The extra complexity of the decision rule does require that two addition nominal probabilities, relating to the minimum probability of making correct decisions under the null and alternative hypotheses, are specified. By way of illustration, in the same setting as that described in section 2 (i.e.
A further option which should be considered as a means with which to effectively address the challenge of multiple endpoints is to use a Bayesian decision-theoretic framework, as employed by Stallard et al.
29
and others in the drug context. This involves the specification of a utility function
3.1.2 Trial specification
A further difficulty arising from the use of multiple endpoints is encountered when setting the specific design parameters for the trial. As illustrated in Figure 2, increases in the number of endpoints can correspond to increases to the dimensions of space in which the decision rule is defined. Accordingly, the number of potential decision rules which could be considered can increase. This feature can been seen in the phase II context when comparing the two-stage design of Simon, 25 which accounts for a single endpoint, with its extension to the two-endpoint setting proposed by Bryant and Day. 28 In that case, given a proposed maximum sample size for each stage of the trial, n, the two-endpoint design will have a factor of n2more possible parameterisations than the single-endpoint design. As a result, the task of finding the specific ‘best’ parameterisation becomes more demanding and less amenable to simple, exhaustive search methods. This point is noted by Sill et al., 30 who propose heuristic methods to find good parameterisations of their two-endpoint phase II design.
In the complex intervention setting, the presence of several endpoints will compound this difficulty and lead to more sophisticated optimisation routines being required as standard. The design of any such algorithms will be strongly influenced by the nature of the endpoints considered. Binary endpoints will lead to integer trial design parameters (e.g. the threshold number of observed successes), whereas continuous endpoints will lead to continuous design parameters (e.g. the threshold of a t-test). Optimisation algorithms are typically tailored to specific problem types, 44 and so different methods will generally be required to solve different problem types efficiently. Metaheuristic algorithms such as genetic optimisation, as implemented in the R package ‘rgenoud’, 45 may provide a flexible solution methodology to address this difficulty, requiring only the tuning of algorithm parameters to ensure good performance.
Where a Bayesian decision-theoretic framework is employed, a decision rule does not have to be specified in advance. The aforementioned method of MEU does not require one, 39 instead determining the decision by choosing that which, conditional on the observed data, has greatest expected utility. As a result, when determining the best specification for a trial one will not need to explore different decision rules. The addition of further endpoints will therefore not lead to a more challenging trial design problem, in contrast with some frequentist cases.
3.2 Clustering
Clustering is a common feature of complex intervention trials and may arise with or without cluster randomisation.
46
For example, while the OK-D feasibility trial is individually randomised, the assumption that patient outcomes are independent is questionable. This is due to the fact that, in the intervention arm of the trial, each participant is allocated to one of a limited number of trained research nurses whose role is to provide support in the delivery of the intervention. The study design is summarised in Figure 3.
Clustering within the OK-D feasibility study, where patients are randomised to intervention or control and those within the intervention arm are allocated nurses.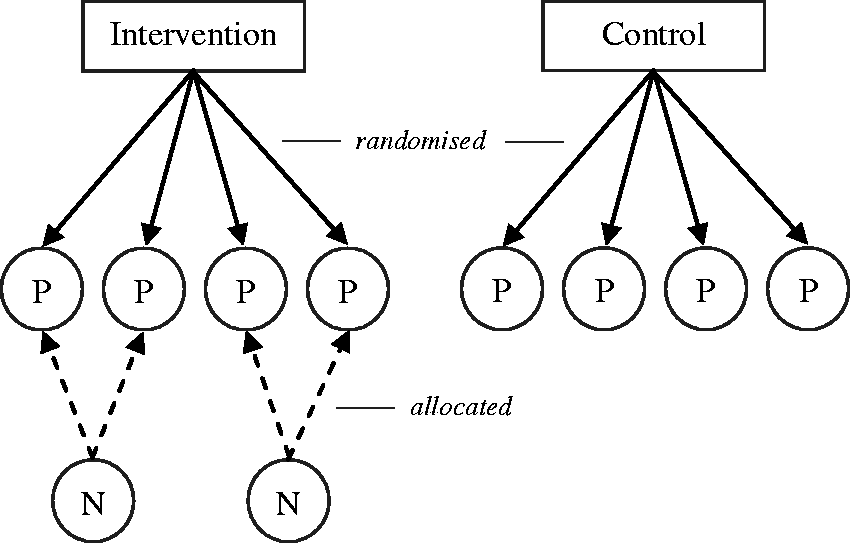
The OK-D study may be described as having an individually randomised, two level, partially nested hierarchical design and is one of many possible scenarios where one or more sources of clustering are present. 17 By partially nested, we refer to the fact that clustering by research nurse is present in only one of the two arms, and by hierarchical we mean that there is a single research nurse per patient. More generally, the relationship between clusters and patients may be hierarchical, cross-classified (where patients are allocated to more than one type of cluster) or multiple-membership (where patients are allocated to more than one cluster of the same type). In terms of the relationship between treatments and clusters, this could be described as partially or fully nested, partially or fully crossed, or a mix of these for trials with more arms. 17 Specifically, nested designs have different clusters in each arm. For example, Schnurr et al. 47 describe a nested trial comparing Prolonged Exposure to Present-Centred Therapy for women with Posttraumatic Stress Disorder, where each therapist delivered only one of the treatments. Crossed designs have different arms associated with the same clusters. 48 Cohen and Mannarino 49 describe one such trial, comparing Cognitive Behavioural Therapy with Nondirective Supportive Therapy for sexually abused children, where therapists delivered both treatments.
In seeking to apply a phase II design to a problem where clustering is present, the simplest approach would be to ignore the clustering and apply the design ‘off-the-shelf’ without any modification. However, this can lead to inaccurate estimates of the type I error rate of any proposed trial 50 with the actual rate being higher than that calculated when designing the trial. As such, this approach would lead to ineffective interventions being taken forward for further evaluation in a phase III trial. A phase II design could be extended to account for clustering by including fixed cluster effects. However, such an analysis would imply a restricted focus on the specific clusters considered in that trial, preventing any generalisation to a wider population. In the case of the OK-D feasibility study, this would correspond to restricting attention to only those nurse therapists participating in the experiment, as opposed to considering the larger population of therapists from which they are ‘sampled’.19,51,52 While it has been argued that this perspective is appropriate in the early phase of development, 51 it is possible to improve the generalisability of the analysis by using random cluster effects rather than fixed. This approach has been recommended to account for clustering in individually randomised trials,46,53 but will lead to a more complex linear mixed effects model.
3.2.1 Complex likelihoods
The hypothesis testing approach typical of phase II trials requires the specification of a test statistic and the derivation of that statistic’s sampling distribution under the null and alternative hypotheses. Given analytical formulae describing these distributions, error rates for any decision rule can then be found by examining their tail areas. This approach is feasible in cases such as those considered by Fleming 22 and A’Hern, 23 where the distribution of the test statistic (a count of binary ‘successes’) is simply the binomial distribution. In multilevel statistical models, as found in trials where clustering is present, statistics such as a mean difference in a linear mixed effects model fitted by maximum likelihood will not necessarily have known analytical sampling distributions,54,55 particularly in our setting where low sample sizes preclude the use of asymptotic results. 56
When analytical results describing the sampling distribution of the test statistic are not available, Monte Carlo simulation may be employed to estimate type I and II error rates.55,57 This involves simulating a number of hypothetical data sets according to a population model which corresponds to either the null or alternative hypothesis and, for each data set, calculating the test statistic. Implementing the proposed decision rule, the resulting action can be compared with the hypothesis used to generate the data and any error, type I or II, counted. This general technique is highly flexible. It can be applied to almost any multilevel structure encountered in practice,58,59 using any proposed statistic in the analysis. However, this flexibility comes at the expense of a computational burden. The Monte Carlo method can require a significant amount of CPU time in order to perform enough simulations to provide an accurate estimate of error rates. The binary nature of both type I and II errors implies that the width of a confidence interval around an estimated error rate will decrease at a rate of
The computational burden of simulations may be reduced through their implementation in efficient programming languages such as C++. However, it has been argued that the resulting lack of transparency and difficulties in interpretation, in comparison to popular statistical programming packages such as R, should be taken into account when considering this option. 60 Alternatively, one may expedite the process of selecting an appropriate sample size by simplifying the problem. This technique is used in the freely available MLPowSim 58 software, which identifies a sensible choice of sample size by calculating the power of a restricted grid of designs, incrementing sample size parameters such as the number of clusters and the number of patients per cluster in large steps. By not considering every possible combination of sample size parameters, precision is sacrificed for speed. In the Stata routine SimSam, 59 the problem is simplified by assuming all but one sample size parameters are known and fixed. Using heuristics to increase the efficiency of the search process, the optimal value of the remaining parameter (e.g. the number of patients per cluster) can be found in a timely manner. An alternative approach would be to use optimisation algorithms which employ surrogate models, such as efficient global optimisation (EGO) 61 and its variants, to search over the full space of sample size configurations. These algorithms rely on fitting models, such as Gaussian processes, to the simulated data obtained over a limited number of initial sample size configurations. Optimisation then takes place over the surrogate model, increasing efficiency as each evaluation now requires a simple calculation as opposed to a full simulation process. As these algorithms and their components have been implemented in R packages 62 and C++ libraries, 63 they can be employed for this purpose without significant difficulty.
The simulation approach may be difficult to implement in cases where ‘nuisance parameters’ are present in the statistical model. This will often be the case where clustering is present. For example, in a fully nested design one would require a value for the ICC to be used in the population model when generating the data at each step. While it has become increasingly common for ICCs to be reported in the results of trials, 64 the early phase context of feasibility and pilot studies implies that little information will be available for the intervention in question. Indeed, gathering information to inform future estimates of ICCs is a common objective of feasibility studies. 8 Thus, calculations of error rates may be dependent on parameter estimates in which there is significant uncertainty. The effect of such uncertainty in ICC estimates on type II error rates and required sample size has been shown to be considerable.65,66 One approach to address this difficulty would be to carry out a sensitivity analysis, using several values of the nuisance parameter covering an appropriate range in order to identify its effect. 55 However, this would further contribute to the computational burden of the simulation approach.
In cases where some information regarding the likely values of nuisance parameters is available, a Bayesian approach would allow for this to be included formally via prior probability distributions. 43 This would fit naturally into the simulation method described thus far, allowing the data generated by the population model to encapsulate uncertainty in the nuisance parameters, leading to more robust estimates of error rates. In the case of ICCs in cluster randomised trials, the use of a prior distribution has been shown to significantly affect both design67,68 and analysis.65,66 In addition to acknowledging uncertainty in parameters, a Bayesian approach will also facilitate the incorporation of information from other sources. Recent methodology has been developed to allow for the weighting given to such prior beliefs to be adaptively changed in response to the data observed in the current trial, 69 where the weighting will decrease as the observed data becomes less commensurate with the historical data. 70 Computationally, the Bayesian approach will require the use of Markov Chain Monte Carlo (MCMC) methods and, as a result, may present difficulties with respect to timely analysis.
3.2.2 Sample size
In addition to leading to complex statistical models, clustered trial designs present difficulties when interpreting the notion of sample size. In phase II designs, sample size is commonly used as a metric with which to compare the quality of any two trial specifications. Typically, the setting of trial parameters is done in such a way as to minimise sample size subject to type I and II error rates remaining within nominal bounds. Trials with clustering, however, will induce further measures to be minimised by the trial designer. For example, the OK-D study involves k research nurses, each of whom has been assigned mpatients. We wish for both k and m to be kept as low as possible whilst ensuring error rates remain within nominal bounds, but these measures are clearly in conflict – reducing one will require increasing the other in order to maintain error rates.
One approach to this problem is to combine the measures into a single weighted combination. This may be achieved through translating each measure to a common scale, such as cost.
71
This would then allow one to focus on minimising cost (subject to constrained error rates). Where such a transformation is not available or appropriate, one may still employ a weighted combination method, although it may be challenging to elicit and represent the preferences of the decision maker(s) in this form. An alternative approach would be to set a limit on one measure, so that the other may be minimised subject to this constraint. For example, one could look for the trial with smallest m such that
3.2.3 Design space
In section 3.1.2, additional complexity in the specification of decision rules was shown to lead to a more difficult optimisation problem due to an increased number of parameters. Similarly, increasing complexity in terms of multilevel structures due to clustering will also require further parameters or dimensions to be considered when searching for optimal trial specifications, 71 and so again it may be beneficial to implement sophisticated optimisation routines rather than exhaustively searching through all possible options. Practically, the impact of increased numbers of design parameters may be limited by bounds on their values. For example, the number of therapists available to deliver an intervention may be fixed, and so when designing the trial one will not have to consider its variation. While such a feature will lead to a simpler optimisation problem, it may also lead to difficulties with regards to parameter estimation and inference.
4 Conclusions and further work
Currently, guidelines for the development and evaluation of complex interventions suggest that early phase experimental work focuses on assessing the feasibility and optimal design of a planned phase III definitive RCT. This contrasts with the drug development setting, where phase II trials are commonly used as a screening mechanism, designed to assess the efficacy of a new treatment and decide if a phase III trial will be worth conducting.
In this paper we have considered how the efficacy of complex interventions could be assessed in the context of current early phase feasibility or pilot studies. With reference to a range of phase II trial designs, challenges to their adaptation to the complex intervention setting have been discussed. The presence of multiple endpoints on which a decision must be based, and the clustering of outcomes in multilevel data structures, have been reviewed in detail. Two recurring themes have emerged. Firstly, the potential benefits of Bayesian methods have been highlighted in the context of decision theoretic approaches to trial design, incorporating uncertainty in trial design parameters and providing robust methods of estimation when only limited numbers of clusters are available. Secondly, we have emphasised the practical need for a sophisticated approach to defining and locating the ‘optimal’ trial specification for a given problem, in order that the best possible trial specification can be determined in a timely and robust manner.
In addition to difficulties arising from multiple endpoints and clustering, there remain several other features which could be explored in future work. One could consider widening the set of decisions of the study from the simple {stop, go} to encompass the refining of the intervention’s components or parameters, 6 or to include the design specification of the planned phase III study in response to feasibility findings. Further details such as the impact of learning curves could be explored, and the appropriate place of efficacy assessment in the larger development and evaluation framework proposed by the MRC 1 should be considered.
Footnotes
Acknowledgements
The authors wish to thank the OK-Diabetes study team (NIHR HTA grant reference 10/102/03) for helpful discussions that shaped the scope of this paper.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Duncan Wilson is funded by a Research Methods Fellowship from the National Institute for Health Research.