
Abstract
Peripheral nerve injury (PNI) often results in persistent functional deficits, and current treatments remain suboptimal. This study developed a tissue-engineered graft by integrating Cdc42-modified bone marrow-derived mesenchymal stem cell (BMSC)-derived exosomes (Exos-Cdc42) with an acellular nerve allograft (ANA) and evaluated its therapeutic potential for nerve regeneration and functional recovery. Exosomes were isolated from BMSCs, and Exos-Cdc42 were generated by transfecting these cells with Cdc42 overexpression vectors. In vitro, Exos-Cdc42 significantly enhanced Schwann cell proliferation, migration, and secretion of neurotrophic factor (BDNF, NGF, CNTF), while upregulating repair-associated markers and downregulating myelination-related markers. In vivo, the combination of Exos-Cdc42 and ANA improved functional recovery of the sciatic nerve, as evidenced by higher sciatic functional index scores and increased muscle weight. Histological analyses demonstrated enhanced axonal regeneration and myelination, characterized by thicker myelin sheaths and larger axon diameters. These findings suggest that Exos-Cdc42 enhance the therapeutic efficacy of ANA by promoting Schwann cell-mediated repair responses, representing a promising strategy for peripheral nerve regeneration.
Highlights
(1) Cdc42-modified BMSC-derived exosomes promote Schwann cell proliferation and migration, which are essential for nerve regeneration. (2) Cdc42-modified BMSC-derived exosomes enhance ANA’s therapeutic efficacy in repairing sciatic nerve defects and promoting functional recovery. (3) Cdc42-modified BMSC-derived exosomes accelerate nerve regeneration, enhance myelination, and improve functional outcomes in peripheral nerve injury models.
Introduction
Peripheral nerve injury (PNI) is a prevalent and debilitating condition that affects a significant number of individuals worldwide each year. 1 The incidence of PNI is increasing due to both traumatic injuries and the aging population, making it an increasingly important public health concern.2,3 Despite advancements in therapeutic approaches, the mechanisms by which these treatments promote nerve repair, especially in cases of extensive or severe injuries, remain incompletely understood. 4
In recent years, therapies utilizing stem cells, particularly applications involving mesenchymal stem cells (MSCs), have garnered considerable attention as potential treatments for PNI.5,6 MSCs, often referred to as “seed cells”, have demonstrated the capability to enhance the repair of peripheral nerve damage by supporting the function of peripheral nerve-related cells.7,8 MSC transplantation has been demonstrated to facilitate nerve regeneration through multiple mechanisms, including transdifferentiation into Schwann-like cells, secretion of neurotrophic factors, modulation of inflammation and immune responses, synthesis of extracellular matrix components, and promotion of angiogenesis.9,10 Despite these promising effects, the clinical application of MSCs is hindered by challenges such as low cell survival rates, limited retention times, reduced post-transplant viability, and complications related to in vitro expansion. 11
Exosomes, small extracellular vesicles secreted by living cells, have emerged as crucial mediators of intercellular communication. Their significant roles in tissue repair, immune regulation, and cell regeneration have been well documented.12,13 Recent evidence suggests that MSC-derived exosomes may facilitate PNI repair by promoting the proliferation of Schwann cells (SCs) and nerve regeneration.7,14 Specifically, bone marrow-derived MSCs (BMSCs) have been implicated in the enhancement of sciatic nerve myelination and regeneration following PNI.15,16 BMSC transplantation has been demonstrated to promote axonal growth and myelin formation, contributing to the restoration of motor function.17,18 However, the role of exosomes derived from BMSCs in PNI repair remains underexplored, and the underlying mechanisms are not yet fully understood.
Cell division cycle 42 (Cdc42), a key regulator of cell morphology and movement, has been identified as a prominent component of exosomes derived from stem cells. 19 Cdc42 is well known for its critical role in promoting SC proliferation and migration following PNI, as well as its involvement in the regulation of myelination.20,21 Recent research has also hilighted that Cdc42 exerts an essential effect on the proliferation and migration of neural stem cells, which are pivotal for peripheral nerve repair through stem cell-mediated mechanisms. 22 Moreover, Cdc42 is believed to be actively incorporated into exosomes secreted by stem cells, which serve as vesicular carriers for bioactive molecules. 23 These exosomes can transfer Cdc42 and other regenerative factors to target cells, thereby enhancing SC function and promoting nerve regeneration. 24 Multiple lines of evidence suggest that stem cells-derived exosomes, particularly those overexpressing Cdc42, can significantly accelerate nerve repair by modulating the behavior of SCs and facilitating the regeneration process. 22
Acellular nerve allograft (ANA) is decellularized nerve graft that promotes nerve regeneration by guiding injured nerves toward distal targets and restoring nerve innervation to skeletal muscles.25,26 The graft is produced by removing cellular components from donor nerve tissue, leaving behind the extracellular matrix, which provides structural support for nerve growth. ANA has been shown to enhance nerve conduction velocity in animal models of sciatic nerve injury, promote the expression of neurotrophic factors within the spinal cord, and protect spinal cord neurons from damage.27,28 These combined effects contribute to significant functional recovery and neural repair. Additionally, ANA supports the regeneration of peripheral nerve defects in facial nerve injuries. 29 Recent studies indicate that the addition of exosomes can further augment ANA-mediated repair by modulating SC function, enhancing axonal regeneration and myelination. 30 Thus, ANA offers great potential as a scaffold for nerve regeneration, demonstrating its promise in promoting functional recovery through both structural and cellular support.
Building on these findings, this study aims to explore the potential of Cdc42-modified BMSC-derived Exosomes (Exos-Cdc42) combined with ANA to improve the functional and structural recovery of peripheral nerves following injury, providing new insights into the development of more effective therapeutic strategies for PNI.
Methods
BMSC separation and characterization
The isolation of BMSCs was performed using femurs and tibias from 4-week-old male C57BL/6 mice, following previously established protocols. 31 Briefly, the femurs and tibias were dissected in a sterile environment, and bone marrow was flushed out with cold phosphate-buffered saline (PBS; Solarbio, Beijing, China) utilizing a 25-gauge needle. The collected bone marrow suspension was centrifuged at 2000 rpm for 5 minutes, and the resulting cell pellet was resuspended in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12; Thermo Fisher Scientific, USA) containing 1% penicillin-streptomycin solution (Solarbio) and 10% fetal bovine serum (FBS; Gibco, USA). Cells were seeded into T25 flasks and incubated at 37°C in a humidified atmosphere containing 5% CO2. After 3 to 5 days, the medium was replaced to remove non-adherent cells, and adherent cells were cultured until approximately 80% confluency was reached. The cells were then subcultured for subsequent experiments.
The identity of BMSCs was confirmed through several characterization assays. Initially, BMSCs were assessed for their morphology, which exhibited a typical fibroblast-like appearance, confirming their mesenchymal origin. To assess their differentiation potential, BMSCs were induced to differentiate into osteogenic and chondrogenic lineages. For osteogenesis, BMSCs were cultured in osteogenic differentiation medium (Cyagen, Cat# MUXMX-90021) for two weeks. The cells were then fixed with 4% paraformaldehyde and stained with Alizarin Red S (Beyotime, Shanghai, China), which binds to calcium deposits, indicating osteogenic differentiation. For chondrogenic differentiation, 2 × 105 BMSCs were cultured in chondrogenic medium (Cyagen, Cat# MUXMX-90041) for 21 days. The cells were subsequently stained with Alcian Blue (Sigma) to detect proteoglycan accumulation, a hallmark of chondrocyte differentiation. Differentiated cells were visualized using a fluorescence microscope (Leica, Wetzlar, Germany).
To confirm the identity of the isolated BMSCs, flow cytometry was conducted using the following antibodies: anti-CD29 (1:20, ARG42772, Arigo, China), anti-CD34 (1:50, ab81289, Abcam, USA), anti-CD45 (106 cells using 1 µg, ab10558, Abcam), and anti-CD90 (1:500, ab307736, Abcam) on the CytoFLEX system (Beckman Coulter, USA).
Transfection of BMSCs
BMSCs were seeded in 6-well plates at a density of 2 × 105 cells per well and cultured in DMEM/F12 (Gibco) containing 10% FBS until reaching 70–80% confluency was achieved. The transfection procedure involved mixing 1 μg of Cdc42 overexpression plasmid (pcDNA3.1-Cdc42; GeneChem, Shanghai, China) with 3 μL of Lipofectamine 3000 (Thermo Fisher Scientific) in 200 μL of Opti-MEM (Gibco), followed by a 15-minute incubation at room temperature. In case of the negative control (NC) category, 1 μg of an empty pcDNA3.1 vector was similarly prepared. Dropwise addition of the mixture was performed in each well, followed by incubation of the cells at 37°C for 6 hours. After replacing the medium with fresh DMEM/F12 supplemented with 10% FBS, the cells were cultured for an additional 24–48 hours. Transfection efficiency was confirmed by Western blot analysis and quantitative PCR (qPCR) to evaluate Cdc42 expression levels.
Isolation and characterization of exosomes
Exosomes were extracted from the culture medium of passage 2–6 BMSCs and Cdc42-overexpressing BMSCs using an ultracentrifugation method. Briefly, the collected culture medium was subjected to sequential centrifugation at 300 × g for 10 minutes, 2,000 × g for 30 minutes, and 10,000 × g for 45 minutes at 4°C to remove debris and cells. Subsequently, the supernatant was transferred to an ultraclear tube (Beckman Coulter) and centrifuged at 100,000 × g for 70 minutes at 4°C. The exosome-containing pellet was collected, washed with PBS, and centrifuged again at 100,000 × g for 70 minutes. The morphology of the exosomes was observed using transmission electron microscopy (TEM; Hitachi HT-7700, Japan). Their size distribution was analyzed via nanoparticle tracking analysis (NTA) using the NanoSight NS300 system (Malvern Panalytical, UK). Western blotting was performed to validate the presence of exosome-specific markers, including anti-TSG101 (1:1000, ab125011, Abcam), anti-CD63 (1:1000, ab217345, Abcam), anti-Alix (1:1000, DF9027, Affinity), anti-HSP70 (1:1000, AF5466, Affinity), and anti-CD9 (1:500, ab223052, Abcam). Calnexin (1:2000, ab13504, Abcam), an indicator of endoplasmic reticulum, served as a negative control to exclude cellular contamination. Additionally, the expression levels of Cdc42 in exosomes derived from BMSCs and Cdc42-overexpressing BMSCs were measured by Western blot analysis.
BMSC-derived exosome labeling and absorption assay
Exosomes were labeled with PKH26 (Sigma), a red fluorescent dye, following the manufacturer’s protocol. Briefly, after resuspending BMSC-derived exosomes (BMSC-Exos) in Diluent C solution, they received 5-minute incubation with PKH26 dye at room temperature. The labeling reaction was terminated by adding 10% bovine serum albumin (BSA), and the labeled exosomes were washed with PBS to remove unbound dye. Following ultracentrifugation at 100,000 × g for 70 minutes at 4°C, the labeled exosomes were pelleted and resuspended in PBS for subsequent experiments. SCs, seeded in 6-well plates at a density of 5 × 105 cells per well, were incubated with PKH26-labeled exosomes for 6, 12, and 24 hours at 37°C in a humidified environment with 5% CO2. Following incubation, SCs were subjected to two washes with PBS and subsequently fixed with 4% paraformaldehyde for 15 minutes. DAPI (Sigma) was employed for counterstaining the cells, enabling visualization of the nuclei. Immunofluorescence images were acquired using a Leica fluorescence microscope. The presence of red fluorescent puncta within SCs indicated successful uptake of BMSC-Exos over time.
Culture and treatment of SCs
SCs were purchased from Procell (Cat# CP-M111, Wuhan, China). The SC-specific culture medium (Cat# CM-M111, Procell) was also provided by the company. The cells were seeded in 6-well plates and cultured in a humidified incubator at 37°C with 5% CO2. The culture medium was changed every 2-3 days, and the cells were passaged when confluence reached 70%-80% for subsequent experiments. SCs at passages 1-2 were used in the experiment to ensure the stability and biological characteristics of the cells.
In the exosome treatment experiment, SCs were treated with isolated and purified BMSC exosomes (Exos-NC or Exos-Cdc42) at a specified concentration of 60 µg/mL for 24 hours. The addition and culture of exosomes were carried out under sterile conditions with gentle mixing to ensure homogeneous distribution of the exosomes in the culture medium. After treatment, the cells were analyzed for proliferation and migration abilities, as well as the expression of repair-related genes and proteins, to further investigate the influence of exosomes on SC function and repair phenotype. 30
qPCR
The primers in this research.
Western blot
Cells were lysed using RIPA buffer (Thermo Fisher Scientific) containing a cocktail of protease inhibitors (Roche, Switzerland). Protein concentrations were determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Proteins (30 µg) were separated by SDS-PAGE (Bio-Rad, USA) and subsequently transferred onto PVDF membranes (Millipore, USA). After blocking with 5% non-fat milk for one hour, membranes were incubated overnight at 4°C with primary antibodies, including anti-MBP (1:1000, ab7349, Abcam), anti-JUN (1:500, ab31419, Abcam), anti-SOX2 (1:1000, ab92494, Abcam), anti-Cdc42 (1:1000, ab155940, Abcam), anti-KROX20 (1:1000, ab245228, Abcam), anti-p-c-Jun (1:1000, AF3095, Affinity), anti-GFAP (1:1000, DF6040, Affinity), anti-p75NTR (1:1000, DF6821, Affinity), anti-p38 (1:1000, AF6456, Affinity), anti-p-p38 (1:1000, AF4001, Affinity), anti-AKT (1:1000, AF0836, Affinity), anti-p-AKT (1:1000, AF0832, Affinity), and anti-β-actin (1:2000, ab8227, Abcam). Follwing washing with TBST, membranes received one-hour incubation with HRP-conjugated secondary antibodies (1:10,000, ab6721, Abcam) at room temperature. Protein bands was were visualized using an ECL detection kit (Beyotime), and images were captured with the ChemiDoc MP System (Bio-Rad, USA).
CCK8 assay
The Cell Counting Kit-8 (CCK8) assay was employed for assessing the cellular viability. SCs were seeded in 96-well plates at a density of 2 × 103 cells per well. After treatment with exosomes or control medium, CCK8 reagent (Dojindo, Japan) was added to each well. The plates were incubated for 2 hours at 37°C, and absorbance at 450 nm was measured using a microplate reader (Thermo Fisher Scientific).
EdU assay
The 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay was adopted for monitoring cell proliferation. SCs were treated with EdU (20 µM) for 2 hours, followed by fixation with 4% paraformaldehyde. EdU-positive cells were detected using the EdU Detection Kit (Beyotime) adhering to the protocol provided by the manufacturer. The cells were visualized under a fluorescence microscope (Leica), and the percentage of EdU-positive cells was assessed by counting cells across multiple fields of view.
Cell migration
The Transwell migration assay (Corning, USA) was used to evaluate the migration capacity of SCs. A suspension of SCs (1 × 104 cells per well) was seeded into the upper chamber of a Transwell insert with an 8 μm pore size. The lower chamber contained 600 µL of complete medium to serve as a chemoattractant. After 24-48 hours, the cells that migrated to the lower membrane surface were fixed with 4% paraformaldehyde for 30 minutes and dyed with 0.1% crystal violet (Solarbio) for a 20-minute duration. Cell migration was quantified by counting cells in five random fields using an inverted microscope (Olympus, Japan).
ELISA
Enzyme-linked immunosorbent assay (ELISA) was performed to investigte the levels of neurotrophic factors, including BDNF, NGF, and CNTF, in the culture supernatants of SCs. After 48 hours of treatment, the supernatants were obtained and subjected to a 10-minute centrifugation at 1000 × g to eliminate debris. The concentrations of NGF (E-EL-M0815, Elascience), BDNF (E-EL-M0203, Elascience, Wuhan, China), and CNTF (CSB-E07312m, SUSABIO, Wuhan, China) were determined using their respective ELISA kits according to the manufacturers’ instructions. Optical density (OD) was detected at 450 nm using a microplate reader (Thermo Fisher Scientific). Standard curves were generated using known concentrations of BDNF, NGF, and CNTF, and the concentrations of each factor in the samples were calculated based on these standard curves.
Animals
C57BL/6 mice (eight to ten weeks old) obtained from Vital River (Beijing, China) were used to establish a sciatic nerve injury model. The mice were housed in a specific pathogen-free (SPF) environment at 22-24°C with 55 ± 5% humidity and a 12-hour light/dark cycle. Animals had ad libitum access to food and water throughout the experiment. All experimental operations received approval by the Institutional Animal Care and Use Committee of Inner Mongolia Medical University (YKD202201085, approved on 02/03/2022), in adherence to national and institutional guidelines for animal welfare. Proper management and handling were ensured throughout the study, with all procedures designed to minimize animal suffering.
Experimental groups and surgical procedures
Mice were randomly assigned to six experimental groups using a random number-based allocation method. The sample size of six animals per group was determined with reference to previous similar studies and preliminary experimental observations. The groups were as follows: (1) Autograft: the sciatic nerve gap was repaired with a reversed autologous nerve graft; (2) Platelet-rich plasma (PRP): the gap was bridged using a conduit filled with autologous PRP, prepared by a two-step centrifugation method 25 ; (3) ANA: repaired using ANA alone; (4) ANA + BMSCs: ANA pre-seeded with 1 × 106 BMSCs; (5) ANA + Exos-NC: ANA loaded with exosomes derived from BMSCs transfected with a NC plasmid (Exos-NC); (6) ANA + Exos-Cdc42: ANA loaded with Exos-Cdc42. BMSCs and exosome loading into ANA were prepared according to previously established protocols.32,33 Briefly, exosomes were immobilized by immersing ANA in a 1 mg/mL exosome solution with gentle shaking for 12 hours at 4°C. To enhance exosome retention, the ANA was then incubated with 0.1% gelatin solution for 30 minutes at 37°C before transplantation. BMSCs suspended in 10 µL of complete medium were injected into the ANA at four evenly spaced locations.
Surgical procedures were conducted as previously described.34,35 In brief, anesthesia was induced in mice by intraperitoneally injecting chloral hydrate (100 mg/kg), followed by exposure of the right sciatic nerve under aseptic conditions. A 3-mm segment of the nerve was excised, resulting in natural retraction and the formation of a 5-mm nerve gap. The nerve gap in the experimental groups was treated using various methods, including ANA (with or without BMSCs or exosomes), autografts, or a PRP conduit, depending on the group. The conduits or grafts were anchored to the proximal and distal nerve stumps using 9-0 nylon sutures. Muscles and skin were closed in layers, and animals were maintained on a heated blanket at 37°C until full recovery from anesthesia. Postoperative care included daily monitoring and subcutaneous administration of penicillin (20,000 U/day) for three consecutive days to prevent infection. After 12 weeks, the gastrocnemius muscles from both the injured and intact legs were harvested for gross morphological observation and analysis. Wet muscle weight was measured immediately after dissection, and the muscle weight ratio (injured vs. uninjured side) was calculated to assess muscle atrophy and recovery.
Preparation of chitosan nerve conduits
Chitosan nerve conduits were prepared by electrodeposition as previously described. 36 A 1% (w/v) chitosan solution (Sigma-Aldrich, USA) in 1% acetic acid was deposited onto a stainless-steel needle (radius: 0.5 mm; length: 7 mm) under a constant current of 0.1 A for 30 minutes using a DC power supply (LONGWEI, China). The conduits were air-dried, sterilized with ethylene oxide (Ande Technology, China), and soaked in sterile PBS (Gibco, USA) for 30 minutes before use. Then, a total of 10–15 μL of freshly prepared PRP was injected into the lumen of each conduit using a microsyringe (Hamilton, Switzerland) and incubated at 37°C for 30 minutes (Incubator, Thermo Fisher Scientific, USA) before implantation.
Preparation of ANA
Sciatic nerves were harvested from CD1 (ICR) mice under sterile conditions and decellularized following established protocols. 33 Briefly, the nerves were incubated in 0.05 M Tris-HCl buffer supplemented with protease inhibitors (100 ng/mL of aprotinin, 500 ng/mL of leupeptin, and 600 mg/mL of pepstatin A) at 4°C for 96 hours. Then, nerve digestion was performed using Tris-HCl buffer containing DNase I (1 U/mL) and RNase I (5 ng/mL) plus 3% Triton X-100 (pH 7.4) for 10 hours. After thorough washing with PBS, the nerves were sterilized via γ-irradiation and stored in sterile PBS at 4°C. To evaluate the efficiency of decellularization, the prepared ANA samples were homogenized using a tissue grinder (Scientz-48, Ningbo Scientz Biotechnology Co., China). Genomic DNA was extracted from the homogenized tissues using a commercial DNA Extraction Kit (DNeasy Blood & Tissue Kit, Qiagen, Germany) according to the manufacturer′s instructions. The DNA concentration was measured using an ultra-micro spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific). The residual DNA content in ANA was 39.47 ± 4.45 ng/mg tissue, which meets the generally accepted criterion (<50 ng/mg) for effective decellularization.
SFI assessment
The Sciatic Functional Index (SFI) was used to evaluate the recovery of sciatic nerve function. 37 Mice were trained to walk on a straight-line track apparatus enclosed with transparent acrylic walls (length: 50 cm, width: 10 cm) to ensure consistent movement. The track floor was covered with replaceable white paper, and a dark shelter was placed at the end of the track to encourage the mice to walk straight. Footprints were recorded by dipping the hind feet of the mice in black, non-toxic ink, after which the mice were allowed to traverse the track freely. Footprints were analyzed to measure three key parameters: toe spread (TS, the distance between the first and fifth toes), print length (PL, the distance from the tip of the third toe to the heel), and intermediary toe spread (ITS, the distance between the second and fourth toes). These parameters were examined for both the experimental (injured) and normal (uninjured) hind limbs using digital calipers or image analysis software. The SFI was calculated using the formula: SFI = 109.5 × (ETS–NTS)/(NTS) – 38.3 × (EPL–NPL)/(NPL) + 13.3 × (EITS–NITS)/NITS–8.8, where ETS, EPL, and EITS represent the experimental toe spread, print length, and intermediary toe spread, respectively, and NTS, NPL, and NITS represent the corresponding parameters from the uninjured limb. The SFI ranges from 0 (normal function) to -100 (complete dysfunction). All functional assessments were performed by investigators blinded to group allocation.
H&E and toluidine blue staining
Sciatic nerve segments from the injury site were harvested at designated time points and preserved in 4% paraformaldehyde for 24 hours at 4°C to maintain tissue morphology. After fixation, the samples were dehydrated through a series of ethanol solutions and infiltrated with paraffin wax. Afterward, the samples were embedded in paraffin for hematoxylin and eosin (H&E) staining or in epoxy resin for toluidine blue staining. For H&E staining, 4-µm-thick sections were prepared using a microtome, followed by deparaffinization in xylene and rehydration through a graded ethanol series. Following a 5-minute hematoxylin staining, the sections were rinsed with water and stained with eosin for 1 minute. The sections were then dehydrated in ethanol, cleared in xylene, and mounted with a coverslip using a resinous mounting medium. For toluidine blue staining, the repaired nerves were fixed in 2.5% glutaraldehyde solution for 24 hours at 4°C. The tissues were immersed in 1% osmium tetroxide for 2 hours, dehydrated, infiltrated, and sectioned into 1-µm-thick semi-thin slices. The sections were stained with 1% toluidine blue (Sigma) for 20 minutes, rinsed with distilled water, and mounted on glass slides. H&E-stained sections were used to observe general nerve morphology, while toluidine blue-stained sections were employed to evaluate the regeneration of myelinated nerve fibers. Images were acquired using a Nikon light microscope and analyzed with Image-Pro Plus software. Histological staining, image acquisition, and quantitative analyses were performed by investigators blinded to group allocation.
TEM analysis
For ultrastructural analysis of nerve recovery, sciatic nerve tissues were harvested at designated time points and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 2 hours at 4°C. After fixation, the samples were post-fixed in 1% osmium tetroxide for 1-2 hours, followed by dehydration through a graded ethanol series and finally embedded in epoxy resin. Thin sections (70 nm) were cut using an ultramicrotome (Leica) and stained with 3% uranyl acetate and 1% lead citrate to enhance contrast. The sections were then examined under a TEM (Hitachi HT-7700, Japan) to observe regenerated nerve fibers. Images were captured, and morphometric analysis was performed to assess the extent of myelinated axons, the width of myelin sheath, and axonal diameter.
Immunofluorescence staining
The sciatic nerve tissues were fixed, dehydrated, embedded, and sectioned. After blocking with 5% goat serum in PBS containing 0.3% Triton X-100, the sections were incubated overnight at 4°C with primary antibodies (anti-S100, ab52642, 1:100, Abcam; anti-NF200, 60331-1-Ig, 1:300, Proteintech), followed by incubation with Alexa Fluor-conjugated secondary antibodies (ab150077 or ab150080, 1:500, Abcam). DAPI was used to counterstain the nuclei, and the sections were then mounted with an antifade medium. Images were captured using a fluorescence microscope and analyzed with ImageJ software to assess levels of SC marker (S100) and axonal regeneration (NF200). Histological staining, image acquisition, and quantitative analyses were performed by investigators blinded to group allocation.
Statistical analysis
Data were expressed as the mean ± standard deviation (SD). Statistical comparisons among multiple groups were performed via one-way analysis of variance (ANOVA), followed by pairwise comparisons with Tukey’s post hoc test. A student's t-test was employed for comparisons between two groups. A p-value less than 0.05 was considered statistically significant. Statistical analyses were conducted using GraphPad Prism version 10.2.3 (GraphPad Software, USA).
Results
BMSC isolation and detection
We first isolated and identified BMSCs to ensure their purity and functional characteristics. Optical microscopy revealed that the isolated cells exhibited a typical spindle-shaped morphology, a hallmark of MSCs, thereby confirming their identity (Figure 1(a)). To evaluate the differentiation potential of BMSCs, we induced osteogenic and chondrogenic differentiation. Alizarin Red dyeing revealed calcium deposits, indicating successful osteogenic differentiation, while Alcian Blue dyeing showed blue staining in the cells, characteristic of chondrocytes, thus verifying the trilineage differentiation potential of BMSCs (Figure 1(b)). Flow cytometry was employed to further characterize the BMSCs by analyzing the expression of specific cell surface markers. The isolated BMSCs exhibited positive expression of CD90 and CD29, while showing negative expression of CD45 and CD34, confirming the successful isolation of mesenchymal stem cells (Figure 1(c)). These results demonstrate that BMSCs were effectively isolated and characterized, establishing a robust foundation for their subsequent application in the investigation of sciatic nerve repair. Isolation and identification of BMSCs. (a) Optical microscopy showing the morphology of BMSCs. (b) Alizarin Red and Alcian Blue staining of BMSCs for osteogenic and chondrogenic differentiation, respectively. Alizarin Red staining shows calcium deposition (osteogenic differentiation) and Alcian Blue highlights round chondrocyte-like morphologies (chondrogenic differentiation). (c) Flow cytometry detection of CD29, CD34, CD45, and CD90 expression in BMSCs.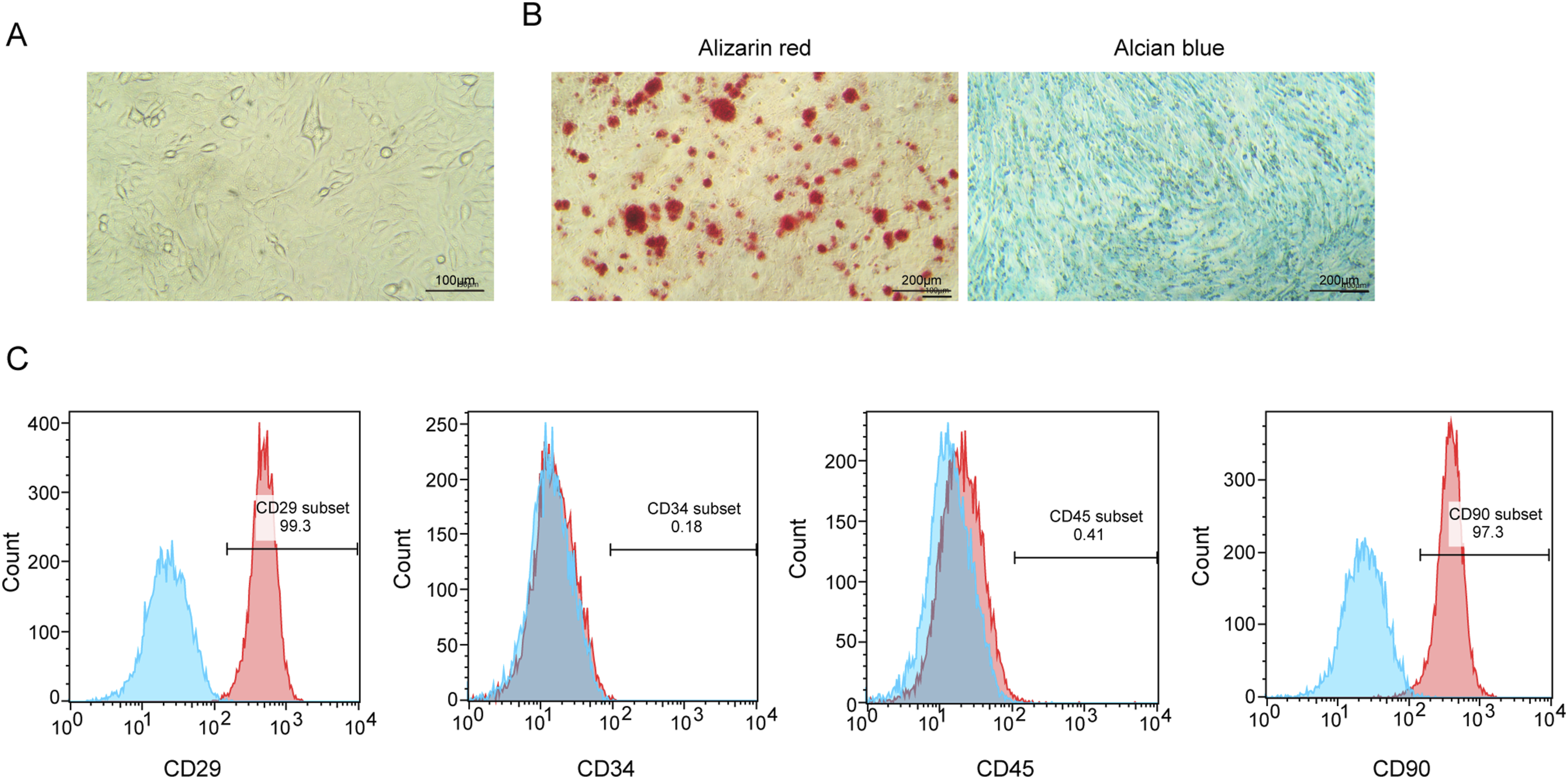
Extraction and identification of BMSC-Exos
The BMSC-Exos were extracted and characterized to confirm their identity and functional properties. TEM images revealed that the isolated exosomes exhibited the characteristic cup-shaped structure with a bilayer lipid membrane, consistent with known exosome morphology (Figure 2(a)). NTA showed that the mean particle size of the isolated exosomes was approximately 148 nm, further confirming their identity (Figure 2(b)). The particle-to-protein ratio was 2.37 × 109 particles/µg. Western blot analysis confirmed the presence of exosomal markers, including CD63, TSG101, and CD9, while the absence of Calnexin indicated high exosome purity and exclusion of contaminating cellular components. In addition to the classical markers CD63, CD9, and TSG101, the expression of Alix and HSP70 was also detected, further validating the identity of the isolated exosomes (Figure 2(c)). Additionally, PKH26-labeled exosomes were incubated with SCs for 6, 12, and 24 hours. Red fluorescent spots were observed within the SCs, indicating successful internalization of the exosomes. A dye-only control (PBS subjected to the same PKH26 labeling procedure without exosomes) showed no detectable red fluorescence, confirming that the observed signals were not due to dye aggregation or micelle artifacts (Figure 2(d)). Extraction and identification of BMSC-Exos. (a) TEM showing the morphology of extracted exosomes. (b) NTA of exosome size distribution. (c) Western blot analysis of exosomal markers (CD9, CD63, TSG101, Alix, and HSP70) and negative control marker (Calnexin). (d) The efficient ability of SCs to internalize PKH26-labeled exosomes.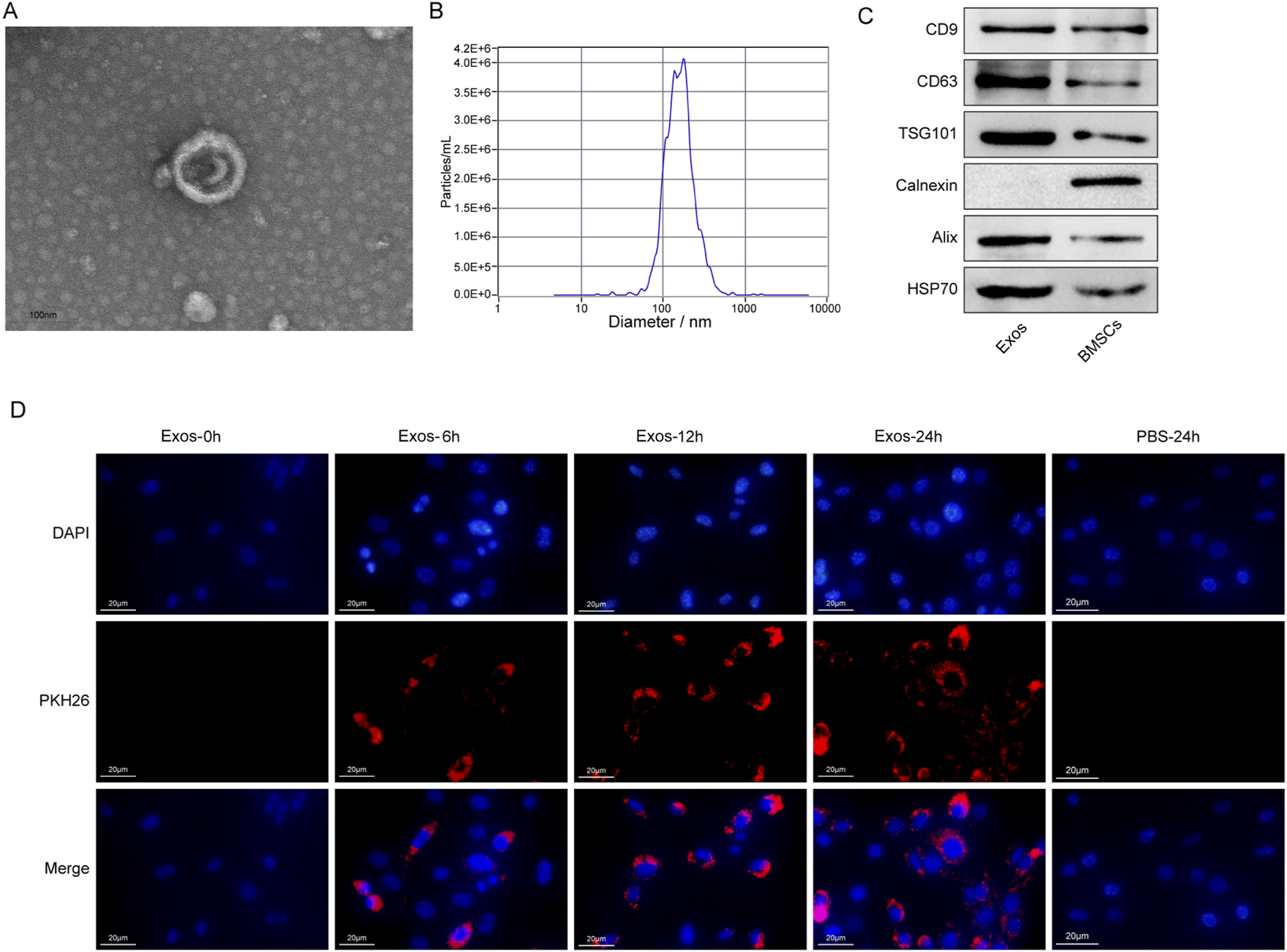
Exos-Cdc42 promoted the proliferation and migration of SCs
First, BMSCs were transfected with a Cdc42 overexpression plasmid, and both qPCR and Western blot analyses showed that Cdc42 mRNA and protein levels were significantly upregulated compared to the control group, confirming successful Cdc42 overexpression in BMSCs (Figure 3(a)–(b)). Next, we isolated Exos-Cdc42 and assessed their impact on the migration and proliferation of SCs. Cdc42 level was elevated in Exos-Cdc42 compared to Exos-NC (Figure 3(c)). SCs treated with Exos-NC demonstrated a notable increase in Cdc42 expression compared to the PBS group, and Exos-Cdc42 further enhanced Cdc42 expression at the protein level (Figure 3(d)). Functionally, the CCK8 assay demonstrated that Exos-NC treatment significantly increased SC viability compared to the PBS group, while Exos-Cdc42 treatment further enhanced SC viability, indicating a stronger proliferative effect (Figure 3(e)). Similarly, the EdU proliferation assay revealed that Exos-NC treatment increased the number of EdU-positive SCs, and Exos-Cdc42 treatment further promoted cell proliferation (Figure 3(f)). Finally, the migration assay revealed that Exos-NC treatment significantly increased the number of migrated SCs compared to the PBS group, with Exos-Cdc42 treatment further enhancing SC migration (Figure 3(g)). Collectively, these results indicate that Cdc42-modified BMSC-Exos promotes SC proliferation and migration. Exos-Cdc42 promoted SC proliferation and migration. (a-b) qPCR and Western blot analysis of Cdc42 mRNA and protein expression in BMSCs. (c-d) Western blot analysis of Cdc42 levels in exosomes (c) and SCs (d) treated with Exos-NC or Exos-Cdc42. (e) CCK8 assay showing SC viability. (f) EdU immunofluorescence staining for SC proliferation. (g) SC migration assay following treatment with Exos-NC or Exos-Cdc42. n = 3. *p < 0.05, **p < 0.01, and ***p < 0.001.
Exos-Cdc42 enhanced the secretory capacity of SCs and activated a repair phenotype
To further evaluate the effects of Exos-Cdc42 on SC repair phenotype, we evaluated the secretion ability and reprogramming process of SCs. First, we examined the expression of neurotrophic factors. Exosome treatment significantly upregulated both protein and mRNA levels of BDNF, NGF, and CNTF in SCs relative to the PBS group. Moreover, Exos-Cdc42 further enhanced the expression of these factors at both proteins and mRNA levels (Figure 4(a)–(b)). Next, we analyzed the activation of genes related to repair and myelination. Treatment with Exos-NC increased mRNA and protein levels of JUN and SOX2, key markers of the SC repair phenotype. Additionally, there was a decrease in the expression of KROX20 and MBP, genes associated with myelination. Exos-Cdc42 significantly upregulated the expression of JUN and SOX2, while downregulating MBP and KROX20 compared to Exos-NC (Figure 4(c)–(d)). The expression of additional repair-associated markers, including p75NTR, GFAP, and phosphorylated c-Jun, was increased following Exos-Cdc42 treatment (Figure 4(e)), further supporting the induction of a repair phenotype in SCs. These findings demonstrate that Exos-Cdc42 not only enhances the secretion of neurotrophic factors but also activates the repair phenotype of SCs. This suggests that Cdc42-modified exosomes may serve as an effective therapeutic tool for PNI by promoting SC-mediated nerve repair and regeneration. Exos-Cdc42 enhanced the secretory capacity of SCs and activated a repair phenotype. (a) qPCR analysis of BDNF, NGF, and CNTF mRNA expression. (b) ELISA analysis of BDNF, NGF, and CNTF protein levels. (c) qPCR analysis of JUN, SOX2, MBP, and KROX20 mRNA expression. (d) Western blot analysis of JUN, SOX2, MBP, and KROX20 protein levels. (e) Western blot analysis of p75NTR, GFAP, and p-c-Jun levels. n = 3. *p < 0.05, **p < 0.01, and ***p < 0.001.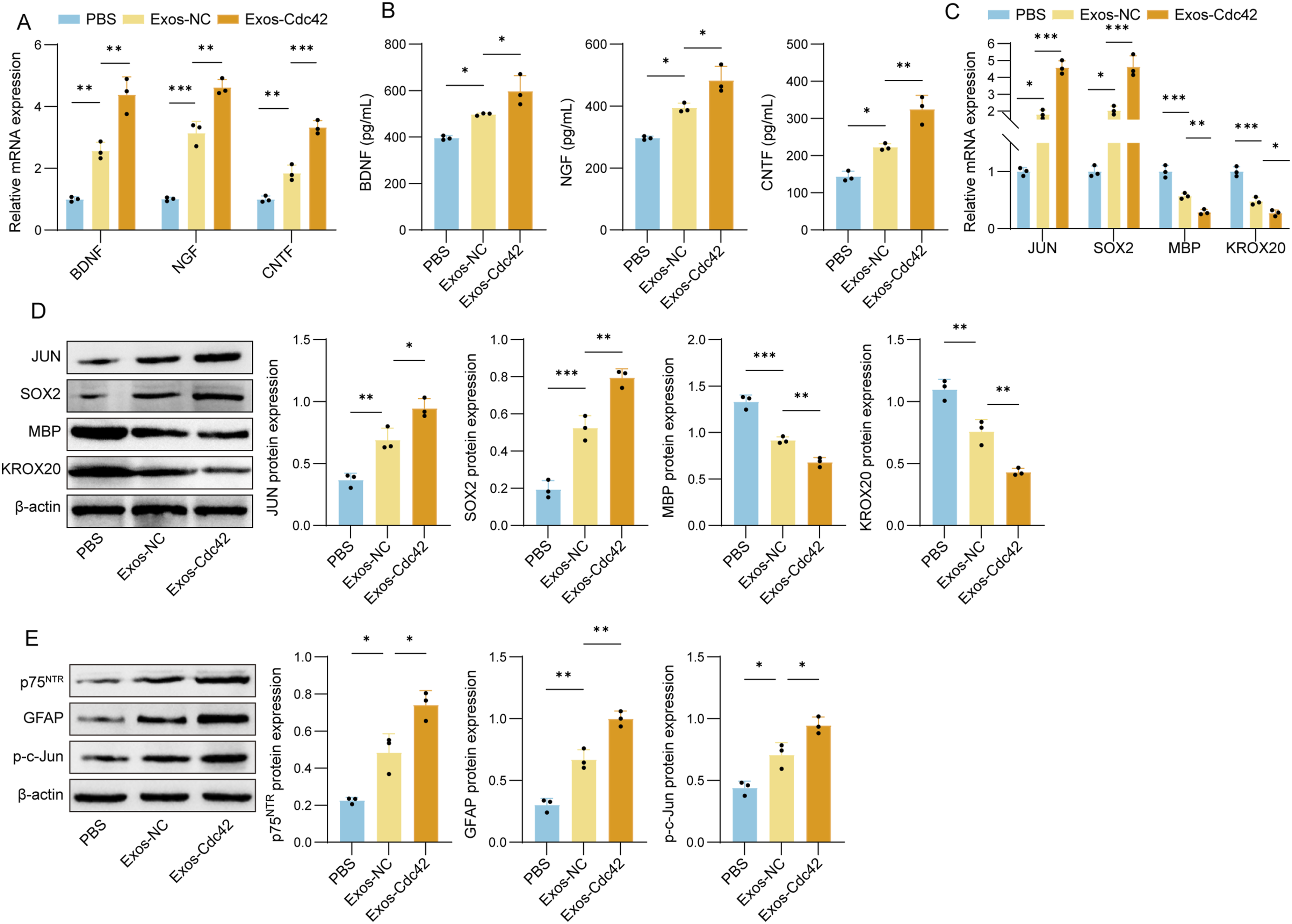
Exos-Cdc42 facilitated the regenerative effects of ANA on sciatic nerve defects
To evaluate the efficacy of Exos-Cdc42 in facilitating PNI repair in vivo, we compared the therapeutic effects of various treatments in mice with sciatic nerve defects. These treatments included autograft nerve, PRP, ANA, ANA combined with BMSCs, ANA loaded with Exos-NC, and ANA loaded with Exos-Cdc42. First, we assessed the SFI to determine the functional restoration of the sciatic nerve. The PRP and ANA groups exhibited lower SFI values compared to the autograft group. Furthermore, the ANA combined with BMSCs and ANA loaded with Exos-NC groups showed significantly improved SFI values compared to the ANA-only group. Notably, the ANA loaded with Exos-Cdc42 group demonstrated the higher SFI values, surpassing those in the ANA loaded Exos-NC group, suggesting that Cdc42-modified exosomes further enhance nerve functional repair (Figure 5(a)). Next, we examined the recovery of muscle weight in the gastrocnemius muscle of the mice. Both BMSCs and their exosomes facilitated the recovery of gastrocnemius muscle wet weight following ANA treatment, with Exos-Cdc42 demonstrating a more pronounced effect compared to Exos-NC (Figure 5(b)). We then analyzed the expression of SC and axon markers, S100 and NF200, which are indicative of myelination and nerve repair. Immunofluorescence results indicated that BMSCs and their exosomes significantly increased the expression of S100 and NF200, with Exos-Cdc42 exhibiting a stronger effect than Exos-NC (Figure 5(c)). Histological examination of the regenerated nerve fibers using H&E staining revealed a dense and orderly arrangement of nerve fibers in the autograft and Exos-Cdc42 groups, which was more pronounced than in the other groups (Figure 5(d)). Toluidine blue staining further confirmed that myelinated nerve fibers were evenly distributed within the regenerated nerve tissue. Both BMSCs and their exosomes promoted the recovery of myelinated nerve fibers, with Exos-Cdc42 demonstrating a more pronounced effect compared to Exos-NC (Figure 5(e)). Finally, electron microscopy revealed that BMSCs and their exosomes significantly increased myelin sheath thickness, the area of myelinated axons, and axon diameter, with Exos-Cdc42 exerting a stronger effect than Exos-NC (Figure 5(f)–(i)). In conclusion, these results suggest that Cdc42-overexpressing BMSC-Exos enhance the repair of ANA in PNI by promoting functional recovery, muscle repair, and the regeneration of myelinated nerve fibers. Exos-Cdc42 facilitated the regenerative effects of ANA on sciatic nerve defects. (a) SFI in the autograft, PRP, ANA, ANA plus BMSCs, ANA plus Exos-NC, and ANA plus Exos-Cdc42 groups. (b) Gastrocnemius muscle gross observation and wet weight analysis. (c) Immunofluorescence staining of S100 and NF200 expression of of regenerated nerves. (d) H&E staining for sciatic nerve repair. (e) Toluidine blue staining for the density of myelinated nerve fibers in regenerated nerves. (f-i) Electron microscopy of the axon diameter, myelin sheath thickness, and myelinated axon area of regenerated nerves. n = 6. *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
PNI is a neurological condition that poses a significant threat to patients, often resulting in severe and long-term functional and physiological disabilities. 38 SCs play a vital role in nerve repair by promoting axonal regeneration and remyelination, but their regenerative potential is frequently insufficient in case of large or severe injuries.39,40 BMSCs have emerged as a promising source of exosomes, which act as carriers of bioactive molecules that regulate the behavior of target cells. Notably, BMSC-Exos have demonstrated the ability to modulate SC proliferation, migration, and secretion of neurotrophic factors, essential processes for nerve regeneration.17,41 This study showed that Exos-Cdc42 enhanced these functions, resulting in improved structural and functional restoration in a mouse model of injured sciatic nerve. Furthermore, the combination of Exos-Cdc42 with ANA produced synergistic effects, highlighting their potential clinical application.
BMSC-Exos play a central role in modulating SC behavior delivering a diverse array of bioactive compunds, including miRNAs, lipids, and proteins, which influence SC proliferation and migration.42,43 Previous studies have shown that BMSC-Exos enhance SC proliferation and migration by activating signaling pathways such as PI3K/Akt and MAPK/ERK, which regulate cytoskeletal remodeling and cell cycle progression.44,45 In this study, Cdc42 was found to play a critical role in promoting SC repair phenotypes and enhancing nerve regeneration, providing a targeted strategy for exosome-based therapies. Exos-Cdc42 significantly improved SC proliferation, migration, and neurotrophic factor secretion by upregulating repair-associated genes (JUN, SOX2) and downregulating myelination-related genes (MBP, KROX20). These findings align with prior studies showing that Cdc42 activation facilitates SC responses to injury through pathways such as PI3K/Akt and p38 MAPK. 46 Exosome-mediated intercellular communication has emerged as a key mechanism in tissue repair and regenerative medicine. Accumulating evidence indicates that exosomes can deliver functional proteins, lipids, and regulatory RNAs to recipient cells, thereby modulating cellular behavior and microenvironmental responses. Recent studies have demonstrated the potential of exosome-based delivery systems in enhancing tissue regeneration, including nerve repair, by improving cell survival, migration, and functional recovery. 47 In addition, exosomes have been shown to play critical roles in intercellular signaling in pathological contexts such as cancer, where they regulate cell proliferation, immune responses, and microenvironment remodeling. 48 In this context, the present study extends current knowledge by demonstrating that Cdc42-modified BMSC-derived exosomes, when combined with acellular nerve allografts, can further enhance Schwann cell-mediated repair responses and functional recovery. These findings support the concept that engineering exosomal cargo represents a promising strategy to optimize regenerative outcomes. Moreover, following nerve injury, SCs transition from a pro-myelinating state to a repair phenotype, characterized by upregulation of JUN and SOX2 and transient downregulation of myelination-related genes.49,50 This shift supports early axonal regeneration. As regeneration progresses, SCs gradually revert to a pro-myelinating state during later stages of axonal regrowth and reinnervation. 51 Therefore, the reduced expression of myelination markers observed in this study likely reflects an early repair phase rather than impaired remyelination. Notably, while previous research has highlighted the general benefits of BMSC-derived exosomes in nerve repair, 30 our study is among the first to integrate Cdc42 overexpression, demonstrating enhanced functional recovery (SFI) and structural repair compared to controls. This builds on earlier findings that Cdc42 regulates SC cytoskeletal remodeling and reprogramming. 22 Our results not only validate the therapeutic potential of Cdc42-modified exosomes but also provide a foundation for future studies to explore their molecular cargo and clinical scalability.
The ability of SCs to secrete neurotrophic factors such as BDNF, NGF, and CNTF is crucial for supporting neuronal survival, axonal growth, and remyelination. 52 BMSC-Exos are known to enhance the secretion of neurotrophic factors from SCs, thereby creating a pro-regenerative environment at the injury site. In this study, Exos-Cdc42 treatment significantly increased the secretion of CNTF, NGF, and BDNF from SCs. This enhancement can be attributed to the molecular cargo of Exos-Cdc42, which includes bioactive molecules that activate the MAPK/ERK and PI3K/Akt pathways, as well as upregulate repair-related genes such as SOX2 and JUN.20,45,53 Consistent with this, we observed increased phosphorylation of AKT and p38 following Exos-Cdc42 treatment (Supplemental Figure 1), further supporting the involvement of PI3K/AKT and MAPK signaling pathways in mediating SC responses. These pathways and genetic changes reprogram SCs into a more regenerative phenotype, enhancing their capacity to repair damaged axons. These results suggest that BMSC-derived exosomes, especially those modified by Cdc42, act as key regulators of the neurotrophic microenvironment, thereby accelerating axonal repair and remyelination.
BMSC-Exos have been shown to enhance nerve regeneration in vivo by modulating SC function and supporting axonal growth and myelination. 54 In this study, we observed that Exos-Cdc42 further amplified these effects when combined with ANA, resulting in superior functional and structural outcomes compared to ANA or PRP alone. ANA serves as a stable scaffold that supports axonal regrowth and guides regenerating nerves, while PRP primarily provides transient growth factors. The combination of Exos-Cdc42 with ANA leveraged the structural support of the graft and the bioactive properties of the exosomes, as evidenced by increased expression of S100, an SC marker indicative of enhanced SC activation and myelination. Exos-Cdc42 treatment significantly enhanced the regenerative capacity of ANA, as reflected in improved functional recovery (higher SFI scores) and more robust structural regeneration, including thicker myelin sheaths and increased axonal diameters, compared to ANA alone. These findings highlight the dual role of BMSC-Exos: first, as direct modulators of SC behavior, and second, as enhancers of the regenerative microenvironment.43,55 Mechanistically, this effect can be attributed to the delivery of Cdc42 and other bioactive molecules, which activate cytoskeletal remodeling and signaling pathways to accelerate axonal regeneration and remyelination.20,56
Despite the promising outcomes of this research, several limitations must be acknowledged. First, although BMSC-derived exosomes, particularly those modified by Cdc42, showed significant therapeutic effects, their molecular cargo remains to be fully characterized. Identifying the specific miRNAs, proteins, or lipids responsible for SC modulation would provide a deeper understanding of their mechanisms. Second, the study was conducted using a mouse model; therefore further research in larger animal models is necessary to confirm the translatability of these findings to human clinical applications. Third, several key challenges remain for clinical translation, including the scalability and standardization of exosome production, potential immunogenicity of allografts and exosome-based therapies, in vivo biodistribution and clearance of exosomes, and long-term safety. These factors may influence therapeutic efficacy and clinical applicability. Consequently, future studies should focus on optimizing exosome production while systematically evaluating biodistribution, immunogenicity, and long-term toxicity, particularly in large-animal models. Fourth, the release kinetics, in vivo retention, and biological activity of exosomes within ANA were not directly evaluated in this study. However, the enhanced functional and histological recovery observed in the ANA + Exos-Cdc42 group suggests effective local delivery. Previous studies have shown that biomaterial scaffolds can prolong exosome retention and sustain release; nevertheless, this remains a limitation that warrants further investigation. Fifth, although Exos-Cdc42 exhibited enhanced therapeutic effects compared to control exosomes, the detailed molecular cargo responsible for these effects was not comprehensively characterized. Exosome-mediated intercellular communication is known to depend on a complex repertoire of proteins and RNAs. Therefore, it remains unclear whether the observed benefits are solely attributable to Cdc42 or also involve secondary alterations in exosomal cargo. Future studies incorporating transcriptomic and proteomic profiling will be necessary to further elucidate the underlying mechanisms. In addition, the 5-mm sciatic nerve gap used in mice represents a commonly adopted short-gap model constrained by anatomical limitations and widely used to evaluate nerve regeneration. 57 However, it does not adequately reflect critical-size defects encountered in clinical settings. Larger defects (≥10 mm), typically established in rat models, may better approximate these conditions. Therefore, these findings should be interpreted as proof-of-concept, and further validation in larger-gap and larger-animal models is warranted.
Conclusions
This study highlights the therapeutic potential of BMSC-Exo, particularly those modified with Cdc42, in enhancing SC-mediated nerve repair. As demonstrated, Exos-Cdc42 significantly activated the SC repair phenotype, promoting SC proliferation, migration, and neurotrophic factor secretion. These effects resulted in improved structural and functional recovery in a sciatic nerve injury model (Figure 6). A neural transplant combining tissue engineering and gene therapy—Exos-Cdc42 incorporated with ANA—was experimentally developed and demonstrated superior therapeutic effects compared to other commonly used tissue engineering approaches. Its therapeutic efficacy showed comparable outcomes to autologous nerve grafts in certain aspects, suggesting its potential as a promising strategy for peripheral nerve repair. A schematic diagram illustrating how Exos-Cdc42 promotes ANA to bridge sciatic nerve defects. This figure was created by the authors using Microsoft PowerPoint (with SciFig plugin).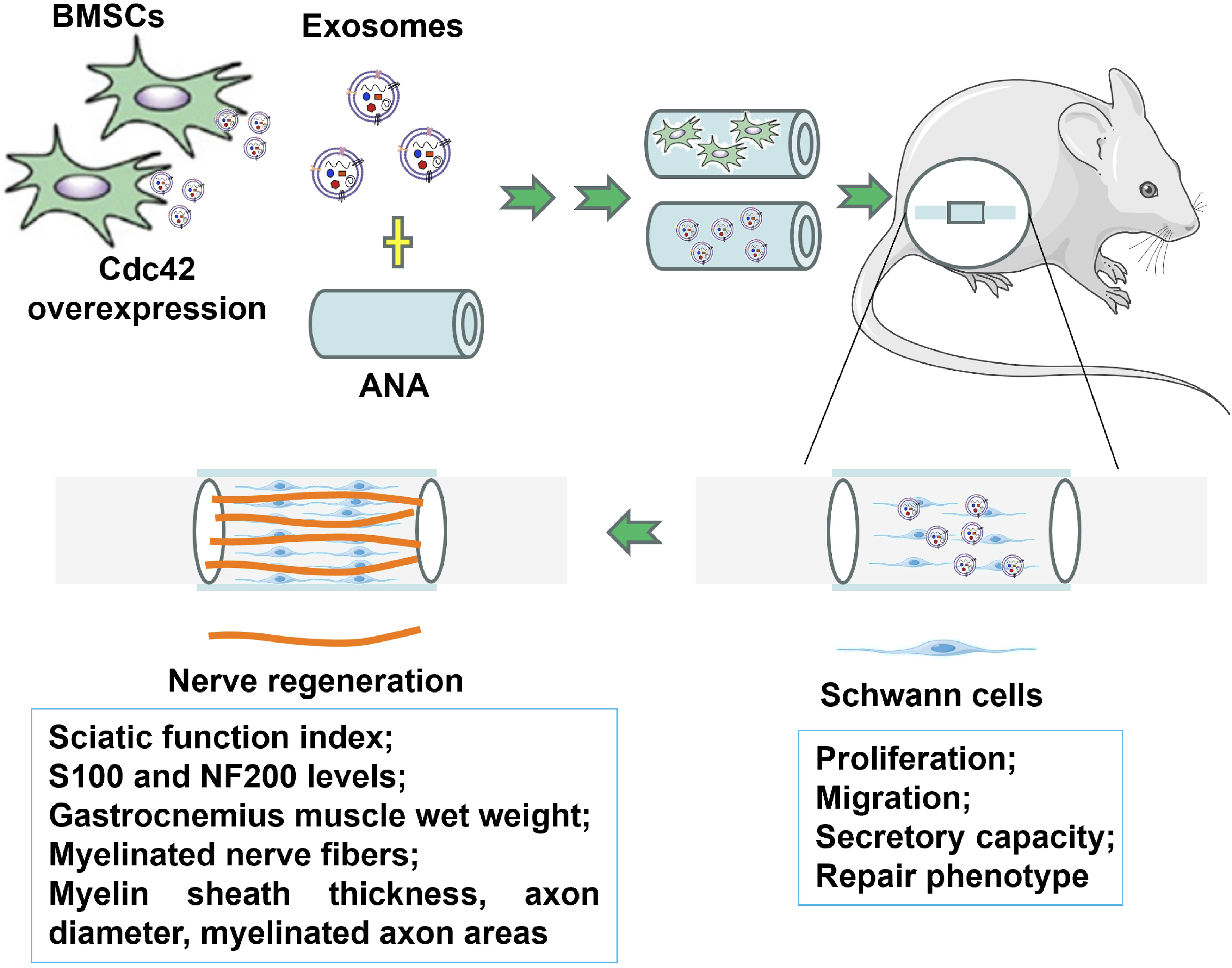
Supplemental material
Supplemental material - Cdc42-Modified BMSC-Derived exosomes promote acellular nerve allografts to bridge sciatic nerve defects
Supplemental material for Cdc42-Modified BMSC-Derived exosomes promote acellular nerve allografts to bridge sciatic nerve defects by Xin Sui, Guangming Dai, Bo Feng in Cell Transplantation
Footnotes
Ethical considerations
All experimental operations received approval by the Institutional Animal Care and Use Committee of Inner Mongolia Medical University (YKD202201085, approved on 02/03/2022), in adherence to national and institutional protocols for animal welfare.
Author contributions
Bo Feng guaranteed the integrity of the entire study, designed the study and defined the intellectual content. Guangming Dai designed the literature research. Xin Sui and Guangming Dai performed experiment, collected the data, and analyzed the data. All authors reviewed the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was sponsored by Science Foundation of AMHT (2022YK027, recipient: Bo Feng).
Declaration of conflicting interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability Statement
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of Generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors did not use any AI-assisted technology.
Supplemental material
Supplemental material for this article is available online.
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.