
Abstract
Objective:
The aim of this study was to explore the potential role of microRNA (miR)-133a, the nucleotide-binding oligomerization domain, leucine-rich repeat family pyrin domain-containing 3 (NLRP3) inflammasome and Sirtuin1 (SIRT1) in the effects of electroacupuncture (EA) on disuse muscular atrophy in mice.
Methods:
C2C12 cells were analyzed for protein expression of NLRP3, SIRT1, myogenin (MyoG) and myosin heavy chain protein (MyHC) after being transfected with a miR-133a-1 mimetic or miR-133a-3p inhibitor. Disuse muscular atrophy was induced in mice by tail suspension, and the mice either remained untreated (DA group) or received EA (frequency 20 Hz, intensity 1 mA, 15 min per day). The morphology of skeletal muscle was measured by muscle/body weight ratios, muscle fiber cross-sectional area (CSA) and expression of Atrogin-1 and Muscle RING finger-1 (MuRF1). miR-133a, NLRP3, apoptosis-associated speck-like protein (ASC), caspase-1 and SIRT1 were detected sequentially. Differentiation of skeletal muscle was examined by measurement of MyoG and MyHC.
Results:
In our in vitro experiments, overexpression of miR-133a downregulated the expression of NLRP3 and SIRT1, and upregulated the expression of MyoG and MyHC. In our in vivo experiments, EA effectively ameliorated muscle wet weight and fiber CSA in the mouse model of disuse muscular atrophy, and decreased Atrogin-1 and MuRF1. EA also significantly increased the expression of miR-133a, inhibited the expression of NLRP3, ASC, caspase-1 and SIRT1, and increased the expression of MyoG and MyHC.
Conclusions:
These results indicate that EA-induced reductions of disuse muscular atrophy may be related to miR-133a. First, miR-133a may inhibit NLRP3 inflammasome activation to reduce muscle loss. Second, miR-133a may regulate SIRT1 expression to induce skeletal muscle differentiation and potentially promote muscle tissue recovery.
Introduction
Disuse atrophy of skeletal muscle is a common disease with multiple causes, which can have a series of adverse effects on the daily life and rehabilitation of patients. 1 Thus, exploring effective treatments for skeletal muscle atrophy is of great potential clinical significance. At present, the treatment of muscle atrophy is mainly through two perspectives: namely decreasing protein consumption,2,3 and facilitating tissue regeneration. 4 Electroacupuncture (EA) is widely used for the treatment of musculoskeletal disorders and skeletal muscle fatigue, and can effectively antagonize the atrophy of skeletal muscle. 5 However, the mechanisms underlying the effect of EA on muscle atrophy is still unclear. Recent studies have confirmed that the amelioration of skeletal muscle atrophy by EA is closely related to changes in microRNAs (miRNAs), 6 for example, miR-133a. 7
miR-133a is a muscle-specific miRNA that is mainly expressed in skeletal muscle and myocardium. It can regulate gene expression post-transcriptionally, 8 and forms a complex biological regulatory network with related transcription factors and signal protein kinases. 6 Recent studies have reported that miR-133a can regulate the expression of nucleotide-binding oligomerization domain, leucine-rich repeat family pyrin domain-containing 3 (NLRP3).9,10 NLRP3 is a well-known inflammasome that is contained within the muscle and is an important molecule in the inflammatory response. 11 Muscle atrophy attributed to the rate of muscle protein breakdown is outpaced by the rate of protein synthesis, and the increase in muscle protein breakdown is related to inflammation. 12 Activation of the NLRP3 inflammasome induces an inflammatory response and elicits catabolic muscle responses that result in muscle loss and atrophy. 2 Previous studies have indicated that EA can exert a protective effect on skeletal muscle by attenuating inflammation.13,14 However, few studies have confirmed the relationship between miR-133a and NLRP3 in EA treatment of disuse atrophy.
Disuse-atrophied muscle will typically restore its mass through regeneration and differentiation. 15 The process of skeletal muscle differentiation is controlled by myogenin (MyoG) and myosin heavy chain protein (MyHC), which play crucial roles in the regulation of muscle mass. Atrophy of skeletal muscle cannot be repaired and may even be aggravated if the differentiation function of the skeletal muscle is disturbed. miR-133a is involved in regulating the growth and development of skeletal muscle, including the differentiation of muscle cells. Shin et al. and Koltai et al. demonstrated that sirtuin 1 (SIRT1) is a downstream transcriptional target of miR-133a.16,17 SIRT1 is a deacetylase that is dependent on nicotinamide adenine dinucleotide (NAD+), which is a key node in the intracellular signal transduction network and involved in mediating various physiological processes of the body. 10 SIRT1 controls the differentiation of mesenchymal stem cells and myogenic progenitors,18,19 implying that SIRT1 might serve as an important target to promote differentiation of cells.
Overall, muscle atrophy may be caused by inflammation resulting in protein decomposition consumption and inadequate myocyte differentiation.12,20 EA can ameliorate muscle atrophy by regulating miR-133a, and miR-133a is associated with both inflammatory related molecules (NLRP3 inflammasome)21,22 and a target molecule of differentiation (SIRT1).16,17 Therefore, it may be that EA affects NLRP3 and SIRT1 expression by regulating miR-133a to ameliorate muscle atrophy. The aim of this study was to preliminarily investigate the relationships between miR-133a, NLRP3 and SIRT1 in EA treatment of disuse muscular atrophy in a mouse model.
Methods
Animals and interventions
Thirty C57BL/6 male wild-type mice (10 weeks old) were weighed and then randomly assigned to three groups: control group (C; n = 10), untreated disuse atrophy group (DA; n = 10) and disuse atrophy group treated with EA (DE; n = 10). Mice were housed in standard cages under 12 h light/dark cycles with ad libitum access to food and water. After the mice had been acclimatized for 2 weeks, the experiments commenced. The present study was approved by the Animal Research Ethics Committee of Chongqing Medical University in accordance with internationally accepted principles for laboratory animal use and care.
Animal models of tail suspension to unload the hindlimb muscles have been used to simulate the bedridden state in humans and this approach has been widely used to induce muscle disuse atrophy in mice. 23 The hindlimb unloading protocol was performed on mice as previously described. 24 During disuse, the hindlimb muscles of mice in the DE group were fixed, and stainless steel filiform needles (0.25 mm × 13 mm; Suzhou Medical Supplies Factory Co. Ltd, Jiangsu, China) were inserted at GB34 (Yanglingquan) and ST36 (Zusanli) to a depth of 5 mm. The needles were then connected to a 6805C-type EA instrument (Shantou Medical Equipment Factory Co. Ltd, Guangdong, China) and stimulated with continuous waves at 20 Hz frequency and 1 mA intensity (pulse width 0.5 ms). EA was given for 15 min every day for 14 days, following which the mice were reweighed, anesthetized using isoflurane (3% for induction and 1.5% for maintenance) and then euthanized by cervical dislocation. The triceps surae (comprising gastrocnemius lateralis, gastrocnemius medius and soleus) was removed immediately and its wet weight was determined using an electronic balance. Muscle samples were then either fixed in 4% formaldehyde solution or stored at −80°C pending further analysis.
Cell culture and transfection
C2C12 mouse myoblasts provided by the laboratory of Army Medical University were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin solution. C2C12 cells were cultured in six-well plates to achieve 80% density at the transfection date (the next day). Cells were transfected with mmu-mir-133a-1 mimetic to induce miR-133a overexpression (miR-133a group) or mmu-miR-133a-3p inhibitor (inhibition group) or negative control (NC group) with HiTransG A/P transfection reagent (REVG003, Genechem, Shanghai, China) following the manufacturer’s recommendations. Cells were subjected to screening for stable transformants by puromycin (1 µg/µL) 72 h after viral transfection.
To suppress SIRT1 expression, the cells were transfected with 100 nM SIRT1 short-interfering (si)RNAs (siR-SIRT1) or siRNA negative controls (siR-NC; RiboBio, Guangzhou, China) according to the manufacturer’s recommendations. C2C12 cells were cultured in T25 flasks to achieve 45% density the next day. Then, the medium was changed to fresh penicillin-streptomycin-free DMEM for transfection. Fresh complete DMEM was replaced 8 h after transfection. SIRT1 expression was determined 36 h later via Western blotting.
Hematoxylin-eosin staining
Paraffin sections were successively dewaxed, dehydrated with an alcohol gradient and stained with hematoxylin and eosin (HE) for 5 min at room temperature. A microscopy system (BX53-LV2000, Olympus, Japan) was used for analysis, and the cross-sectional area of the muscle fibers was calculated using Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA).
Western blotting
The muscle and C2C12 cells lysates were treated with radioimmunoprecipitation assay (RIPA) lysis buffer and protein concentrations were quantified using a bicinchoninic acid (BCA) protein assay (P0010S, Beyotime Biotechnology, Shanghai, China). Thirty milligrams of protein were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; 120 V, 80 min) and transferred onto polyvinylidene (PVDF) membranes (300 mA, 120 min). Then, the membranes were blocked in blocking buffer for 2 h and subsequently incubated overnight at 4°C with the following primary antibodies: mouse anti-SIRT1 (1:1000; ab110304, Abcam, Cambridge, UK), mouse anti-MyoG (1:200; sc-52903, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), mouse anti-MyHC (1:200; sc-376157, Santa Cruz Biotechnology), mouse anti-ASC (1:200; sc-514414, Santa Cruz Biotechnology), rabbit anti-NLRP3 (1:1000; 15101 S, Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit anti-caspase-1 (1:1000; ab138483, Abcam) and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:5000; A00227-1, Boster Biological Technology Co. Ltd, Wuhan, China). The membranes were then incubated with corresponding horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit immunoglobulin (Ig)G secondary antibodies (1:3000; ZB-5305 and ZB-5301, Zhongshan Jinqiao Biotechnology Co. Ltd, Beijing, China) for 1 h at room temperature. Electrophoretic blots were analyzed with Quantity One software (Bio-Rad, Hercules, CA, USA).
Real-time PCR analysis
Total RNA from muscle or cell lysates was extracted using TRIzol reagent (R0016, Beyotime Biotechnology, Shanghai, China) and reverse transcribed with a cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Polymerase chain reaction (PCR) was performed with a CFX connect system (Bio-Rad). The PCR cycling reaction conditions were as follows: 94°C for 4 min, followed by 40 cycles at 94°C for 30 s, 55°C for 30 s, 72°C for 30 s and finally 72°C for 5 min. The cycle threshold (Ct) for each gene was normalized to the cycle threshold for GAPDH. The following primers were obtained from Sangon Biotech (Shanghai, China): Muscle RING finger (MuRF)1 forward CTGCCCTGCCAACACAACCTC reverse GCAACGGAAACGACCTCCAGAC; Atrogin-1 forward TTCACAAAGGAAGTACGAAGGA reverse GCTGGTCTTCAAGAACTTTCAG; and GAPDH forward CAAGGCTGTGGGCAAGGTCATC reverse TCTCCAGGCGGCACGTCAG. The detection reagents for miR-133a (TaqMan miRNA reverse transcriptase kit and TaqMan miRNA assays) were purchased from Applied Biosystems (Carlsbad, CA, USA). Reverse transcription and PCR of miR-133a were performed as described previously. 17
Enzyme-linked immunosorbent assay
Supernatants of C2C12 cells and tissue lysates were harvested and enzyme-linked immunosorbent assays (ELISAs) were performed using commercial kits for interleukin (IL)-1β and IL-18 (PI301 and PI553, respectively; Beyotime Biotechnology) per the manufacturer’s instructions. Levels of IL-1β and IL-18 were determined at a wavelength of 450 nm using a multifunction microplate reader (Model 680, Bio-Rad).
Statistical analyses
All data were expressed as mean ± standard deviation (SD). Groups were compared by one-way analysis of variance (ANOVA), with post hoc least significant difference (LSD) tests for pairwise comparisons, using the Statistical Package for the Social Sciences (SPSS) version 19.0 (SPSS Inc., Chicago, IL, USA). Statistical significance was considered with a p-value of less than 0.05.
Results
EA treatment ameliorated disuse muscular atrophy
Modeling of disuse muscular atrophy resulted in a marked decrease in muscle mass and cross-sectional area that were both significantly ameliorated after EA treatment (Table 1 and Figure 1(a), respectively). Disuse atrophy also increased mRNA expression of MuRF1 and Atrogin-1 (Figure 1(b)). Compared with the (untreated) DA group, expression of MuRF1 decreased by 67% and Atrogin-1 decreased by 53% in the DE group, indicating that disuse muscular atrophy can be significantly ameliorated by EA.
Body weight and weight of triceps surae by experimental group.
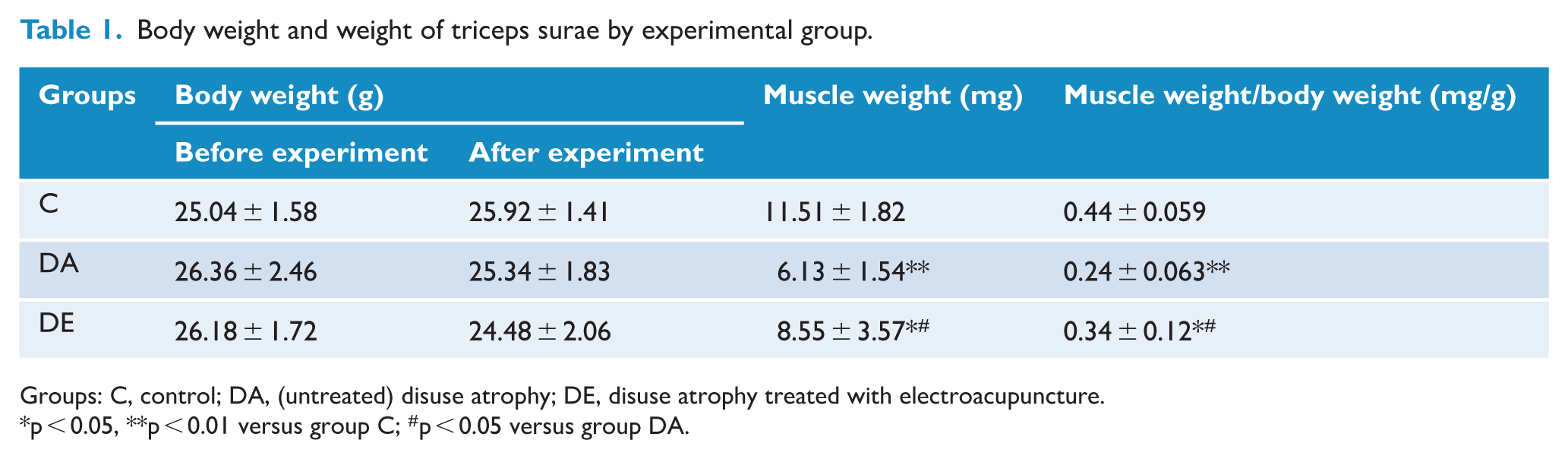
Groups: C, control; DA, (untreated) disuse atrophy; DE, disuse atrophy treated with electroacupuncture.
p < 0.05, **p < 0.01 versus group C; #p < 0.05 versus group DA.
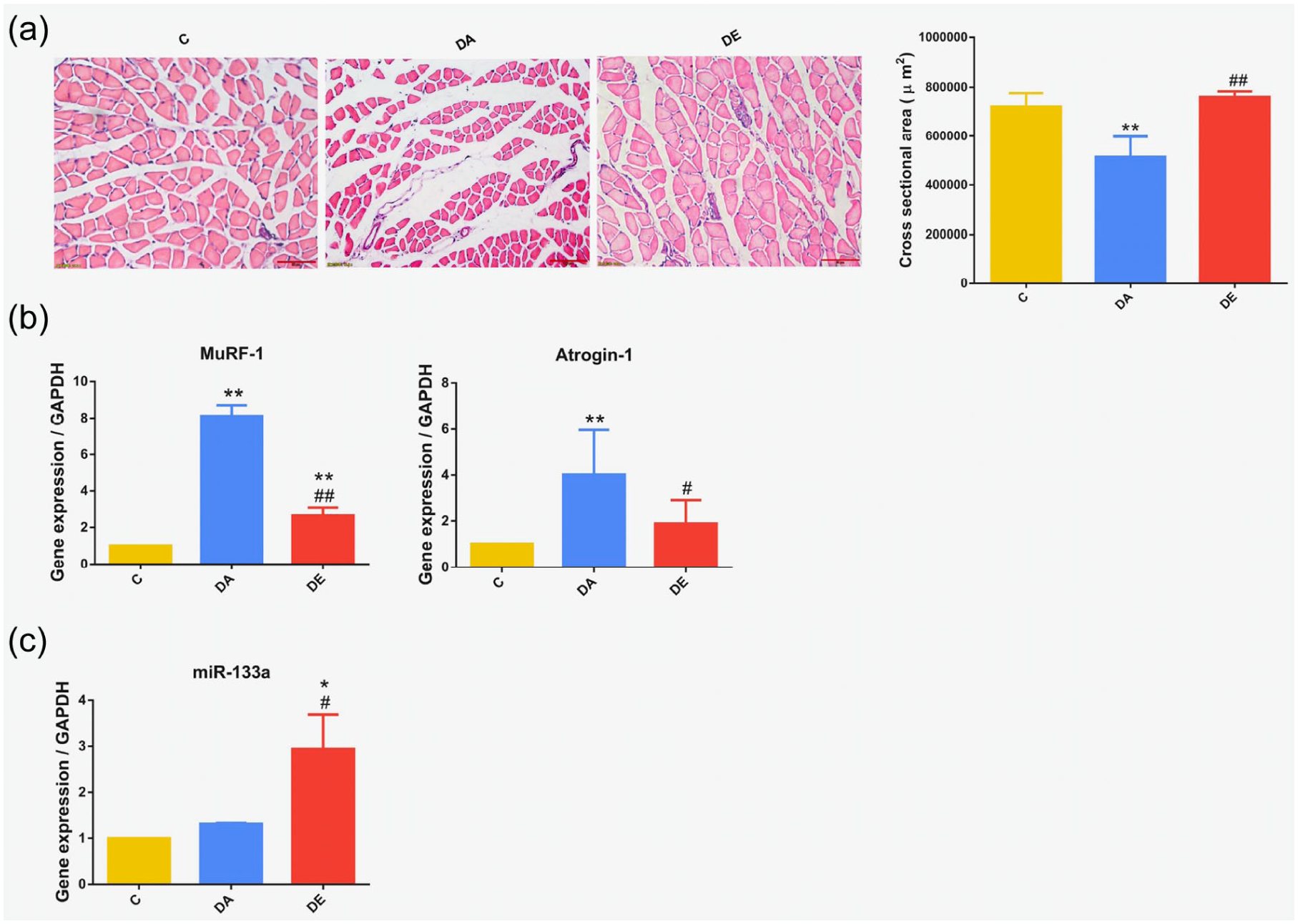
Electroacupuncture (EA) treatment ameliorated disuse muscular atrophy in a mouse model. Groups: C, control; DA, (untreated) disuse atrophy; DE, disuse atrophy treated with EA. (a) Cross-sectional area was measured following hematoxylin and eosin (HE) staining (25.2×, 50 µm). (b) Reverse transcription polymerase chain reaction (RT-PCR) results showing the mRNA expression of Muscle RING finger-1 (MuRF1) and Atrogin-1. (c) EA stimulated microRNA (miR)-133a expression. *p < 0.05, **p < 0.01 versus C group; #p < 0.05, ##p < 0.01 versus DA group.
EA stimulated miR-133a expression
Expression of miR-133a in skeletal muscle was significantly increased after EA treatment (Figure 1(c)). Interestingly, there was no significant difference in the expression of miR-133a in the control group and DA group, while an increase of 194% was noted in the DE group (compared to the control group).
EA inhibited the NLRP3 inflammasome and promotes skeletal muscle differentiation
Figure 2(a) shows protein expression of NLRP3, ASC and caspase-1, which were more highly expressed in muscle atrophy and markedly reduced in muscle treated with EA. The same trend was observed in the levels of inflammatory cytokines. Levels of IL-1β and IL-18 were highly increased in muscle with disuse atrophy and decreased after EA treatment (Figure 2(b)). Meanwhile, SIRT1 expression was significantly reduced, while protein expression of MyoG and MyHC was higher in muscle treated with EA (Figure 2(c) and (d)). We also found that the expression of MyoG and MyHC in the control group was comparatively lower than that in the DE group.
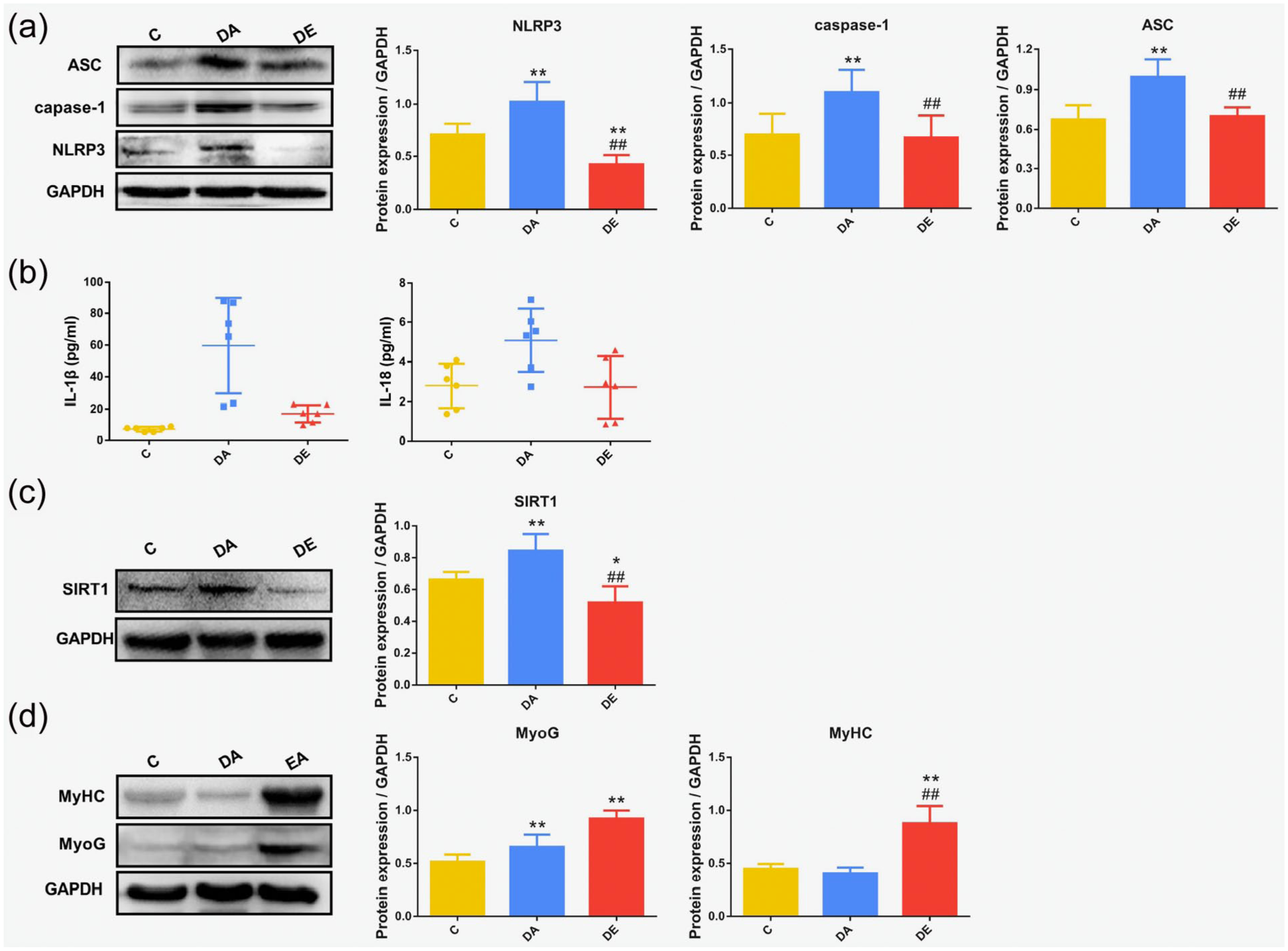
Electroacupuncture (EA) inhibited the nucleotide-binding oligomerization domain, leucine-rich repeat (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome and promoted skeletal muscle differentiation in a mouse model of disuse muscular atrophy. Groups: C, control; DA, (untreated) disuse atrophy; DE, disuse atrophy treated with EA. (a) Western blotting results showing expression of the NLRP3 inflammasome, apoptosis-associated speck-like protein (ASC) and caspase-1. (b) Enzyme-linked immunosorbent assay (ELISA) results showing the levels of interleukin (IL)-1β and IL-18. (c) Western blotting results showing the expression of Sirtuin1 (SIRT1). (d) Expression of myogenin (MyoG) and myosin heavy chain protein (MyHC) as determined by Western blotting. *p < 0.05, **p < 0.01 versus C group; #p < 0.05, ##p < 0.01 versus DA group.
miR-133a inhibited the NLRP3 inflammasome and inflammatory cytokine levels
Our results showed that the expression of NLRP3 was inhibited by the overexpression of miR-133a (Figure 3(a)), followed by ASC and caspase-1 (Figure 3(b)). By contrast, the expression of NLRP3, ASC and caspase-1 (Figure 3(a) and (b)) were significantly increased after miR-133a knockdown. The trend in expression of the inflammatory cytokines IL-1β and IL-18 was consistent with that of NLRP3 (Figure 3(c)).
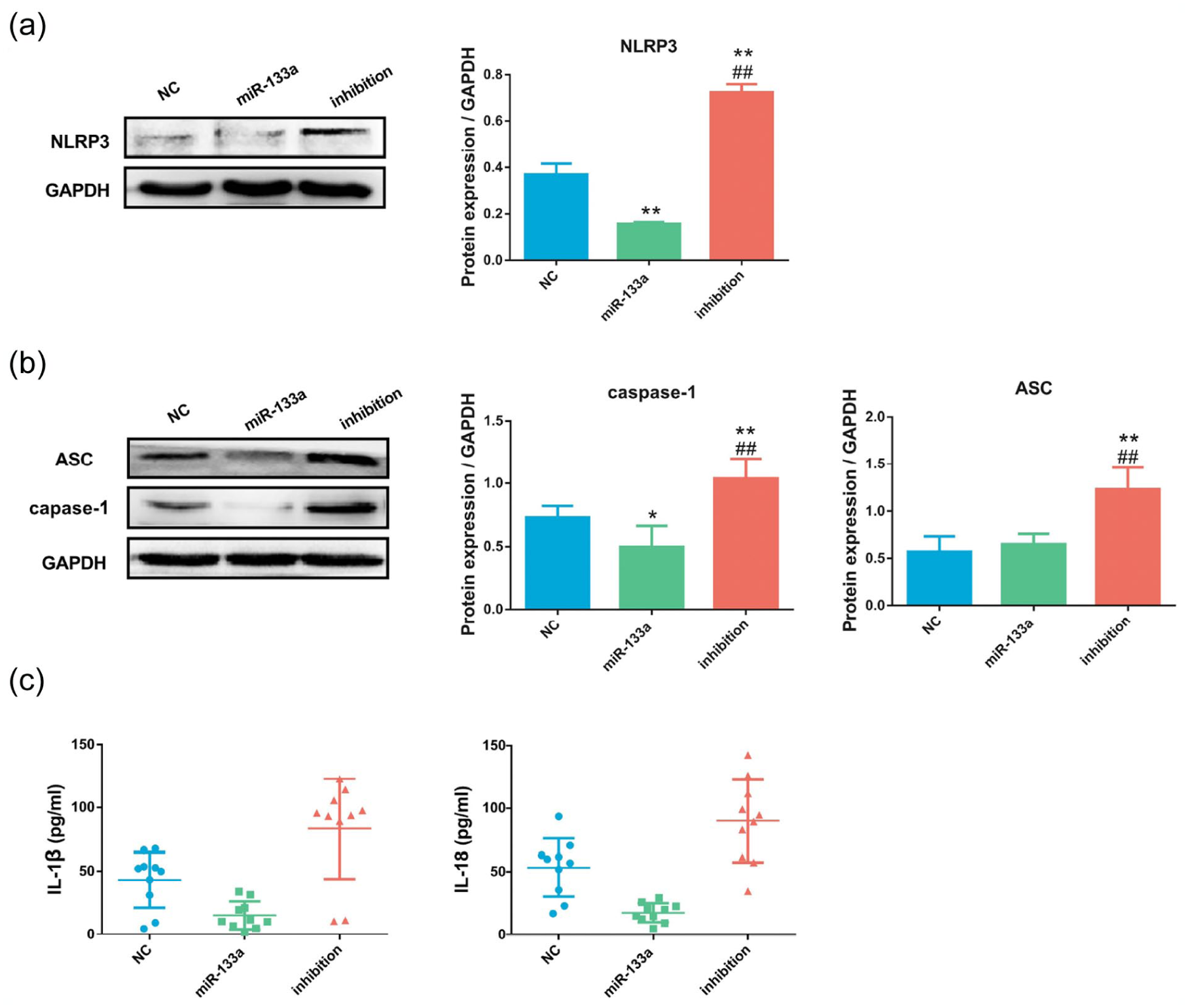
MicroRNA (miR)-133a inhibited the nucleotide-binding oligomerization domain, leucine-rich repeat (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome and inflammatory cytokine levels in C2C12 mouse myoblasts. (a) Western blotting results showing the expression of the NLRP3 inflammasome after viral transfection with mmu-mir-133a-1 mimetic to induce miR-133a overexpression (miR-133a group), mmu-miR-133a-3p inhibitor (inhibition group) or negative control (NC group). (b) Western blotting results showing the expression of apoptosis-associated speck-like protein (ASC) and caspase-1 after viral transfection. (c) Enzyme-linked immunosorbent assay (ELISA) results showing the levels of interleukin (IL)-1β and IL-18 after viral transfection. *p < 0.05, **p < 0.01 versus NC group; #p < 0.05, ##p < 0.01 versus miR-133a group.
miR-133a promoted skeletal muscle differentiation by regulating SIRT1
As shown in Figure 4(a), overexpressing miR-133a led to decreased SIRT1 expression; in contrast, SIRT1 increased after miR-133a expression was inhibited. A positive relationship between miR-133a and the differentiation-related molecules (MyoG, MyHC) of skeletal muscle was observed in the cell experiments, wherein overexpression of miR-133a significantly promoted the protein expression of MyoG and MyHC (Figure 4(b)). Meanwhile, the decrease in SIRT1 promoted MyoG expression in C2C12 cells (Figure 4(c) and (d)). Our results indicate that SIRT1 initiates myocyte differentiation through MyoG, while exhibiting low levels of MyHC expression at this stage (Figure 4(d)).
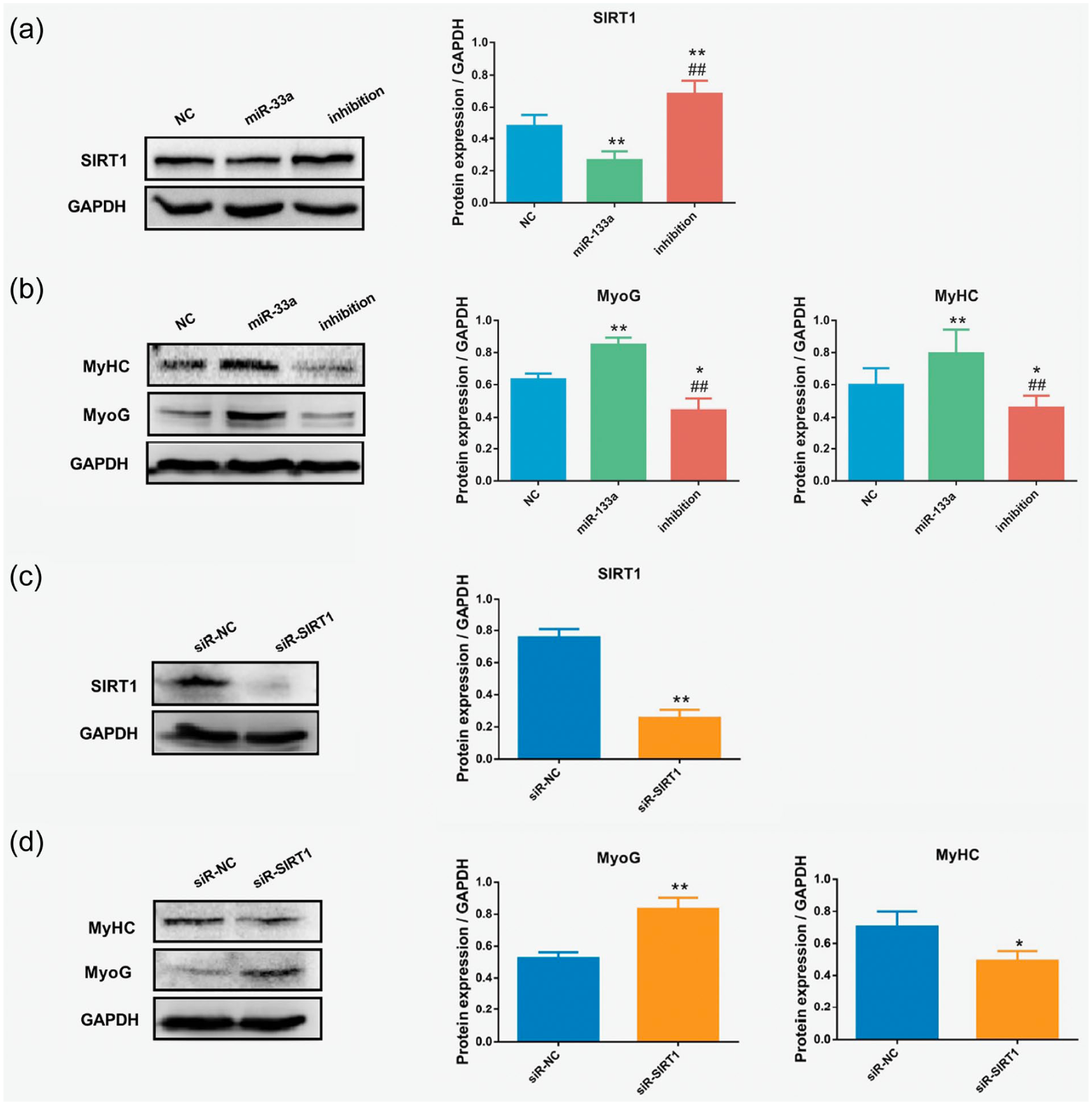
MicroRNA (miR)-133a promoted skeletal muscle differentiation by regulating Sirtuin1 (SIRT1) in C2C12 mouse myoblasts. (a) Western blotting results showing the expression of SIRT1 after viral transfection with mmu-mir-133a-1 mimetic to induce miR-133a overexpression (miR-133a group), mmu-miR-133a-3p inhibitor (inhibition group) or negative control (NC group). (b) Expression of myogenin (MyoG) and myosin heavy chain protein (MyHC) after viral transfection, as determined by Western blotting. *p < 0.05, **p < 0.01 versus NC group; #p < 0.05, ##p < 0.01 versus miR-133a group. (c) Western blotting results showing the expression of Sirtuin1 (SIRT1) after viral transfection with SIRT1 short-interfering (si)RNAs (siR-SIRT1) or negative controls (siR-NC). (d) Expression of myogenin (MyoG) and MyHC after siRNA transfection, as determined by Western blotting. *p < 0.05, **p < 0.01, versus siR-NC group.
Discussion
In the present study, we observed that disuse muscular atrophy significantly reduced the mass and cross-sectional area of skeletal muscle, which is consistent with a previous report. 25 Previous studies have demonstrated that EA can effectively impede muscle atrophy, and also has the capacity to suppress the release of inflammatory cytokines, 13 and facilitate enhanced muscle differentiation. 26 It is well established that muscle atrophy mainly reflects the loss of muscle mass that occurs when protein degradation exceeds protein synthesis. 27 Inflammation is part of the body’s normal response; however, an excessive inflammatory response can stimulate protein-energy depletion and accelerate the breakdown of muscle protein, potentially leading to muscle atrophy. 12 Moreover, activation of inflammasomes and the subsequent release of proinflammatory cytokines (e.g. IL-1β and IL-18) are strongly associated with skeletal muscle atrophy.28,29 Activation of the NLRP3 inflammasome can cleave and recruit apoptosis-associated ASC and caspase-1 to form the inflammasome complex, thus promoting the maturation and secretion of the proinflammatory cytokines IL-1β and IL-18. 30 Increased IL-1β and IL-18 leads to high levels of expression of MuRF1 and Atrogin-1, 28 which are important markers of skeletal muscle atrophy that label muscle proteins as ubiquitin proteins, which are then recognized and degraded by the proteasome. 31 Our data show that inflammasome expression and inflammatory cytokine levels were significantly increased in this mouse model of disuse muscular atrophy, suggesting that the excessive activation of inflammasomes may be the cause of disuse muscular atrophy. Some studies have suggested that EA can reduce the expression of IL-1β and the NLRP3 inflammasome.13,14 In the present study, we found that EA significantly reduced expression of the NLRP3 inflammasome and inflammatory cytokines in disused muscle, while the expression of MuRF1/Atrogin-1 decreased, and muscle mass improved significantly, suggesting that EA ameliorated muscle mass loss by inhibiting the inflammatory response caused by NLRP3 inflammasomes.
When skeletal muscle tends to atrophy, early repair is also one of the key points. Myocytes withdraw from the cell cycle and undergo differentiation to generate new muscle fibers for effective tissue repair. 32 Differentiation of skeletal muscle progenitor cells plays an important role in skeletal muscle repair and regeneration, 33 and EA promotes the recovery of atrophic muscle strength, which is also associated with skeletal muscle differentiation. 26 Studies have identified that SIRT1 expression is downregulated during the differentiation of C2C12 cells, 34 while decreased SIRT1 in stem cells can induce premature differentiation of cells. 35 Interestingly, in our study, MyoG was upregulated while MyHC was downregulated following interference with SIRT1 in C2C12 cells. Numerous reports have demonstrated that MyoG is a hallmark protein of skeletal muscle differentiation, inducing myoblasts to exit the cell cycle and differentiate into myocytes, thereby initiating cell differentiation, 36 and MyHC is mainly expressed at the end of myoblast differentiation. 37 Our results imply that SIRT1 may be primarily involved in the early stage of myocyte differentiation, rather than muscle fiber maturation.
miR-133a is one of the best-characterized muscle-relevant miRNAs and is involved in the regulation of skeletal muscle repair and other processes. 38 miR-133a plays a central regulatory role in cellular differentiation, and overexpression of miR-133a greatly improved multinucleated myotube formation. 39 However, miR-133a did not participate in disuse muscular atrophy, and its expression was not affected by disuse muscular atrophy in an experiment by Chacon-Cabrera et al., 25 but significantly changed with EA treatment, 7 which is consistent with the results of the present study. Moreover, miR-133a-5p decreased significantly after EA pretreatment with an antagonist compared with EA pretreatment alone, 40 demonstrating a possible role of miR-133a in promoting the recovery of muscular atrophy in general, but not in disuse muscular atrophy. In our experiment, the expression of miR-133a was upregulated, and the NLRP3 inflammatory response and SIRT1 expression were inhibited in the atrophied muscle treated with EA, reflecting that there may be an inverse relationship between miR-133a with the NLRP3 inflammasome and SIRT1. Liu et al. showed that overexpression of miR-133a-3p could downregulate the expression of NLRP3, ASC and cleaved caspase-1, and promote cell viability. 41 In addition, Duan et al. found that upregulation of miR-133a inhibits the activation of NLRP3. 22 In another study, miR-133a-3p alleviated cardiomyocyte hypertrophy by inhibiting the activation of the NLRP3 inflammasome in the myocardium. 21 Our in vitro experimental findings provide evidence that miR-133a effectively exerts a negative regulatory influence on the expression of NLRP3. Nevertheless, due to the complexity of inflammatory signaling pathways, the mechanisms underlying the muscle atrophy caused by inflammatory responses may not be associated with miR-133a, necessitating further investigation. Our results showed that muscle atrophy was effectively alleviated after a significant reduction in the miR-133a-mediated NLRP3 inflammatory response following EA treatment. Thus, the present results reveal the role of NLRP3 inflammasome and miR-133a in the EA treatment of disuse muscular atrophy. Other than that, MyoG increased significantly, alongside miR-133a overexpression and SIRT1 inhibition after EA treatment. Our in vivo experiments also confirmed the negative regulatory role of miR-133a on SIRT1, showing that EA can also initiate the skeletal muscle differentiation program and mediate skeletal muscle repair through miR-133a inhibition of SIRT1 expression. Some literature also pointed out that there was a positive regulatory relationship between EA and SIRT1, which was different from our study results, suggesting that the relationship between EA and SIRT1 was not simple. Skeletal muscle differentiation is a complex process, and both SIRT1 and miR-133a play important roles in this process. Prior research has shown that miR-133a inhibits the expression of SIRT1 in cardiomyocytes, and miR-133a/SIRT1 is a key regulator of the terminal differentiation and maturation of cardiac myocytes during cardiac development.16,17 They may have the same mechanism of action in skeletal muscle.
To date, many studies have shown that SIRT1 can negatively regulate NLRP3 but, to determine whether the decrease of SIRT1 affects miR-133a/NLRP3, further experimental studies are needed. Taken together, treatment of atrophied muscle with EA can lead to an attenuated inflammatory response and reduced SIRT1 expression. The lower inflammatory response could reduce muscle loss, while the decrease in SIRT1 may promote the differentiation of skeletal muscle, both of which may contribute to the recovery of atrophied skeletal muscle. Accordingly, we speculate that these may be mediated by miR-133a. The main limitation of the present study is the experimental design, as a control group for EA was not established. Furthermore, an intervention group with miR-133a for in vivo experiments was not arranged, thus our study cannot definitively demonstrate that EA ameliorates disuse atrophy “through” miR-133a. We plan to conduct further investigations to verify and elucidate the mechanism of EA treatment for disuse atrophy.
Conclusion
EA has become a strategy of interest in preventing and ameliorating muscle wasting. The new findings in our study regarding the role of miR-133a in the reduction of disuse muscle atrophy by EA may provide new insights into potential therapeutic strategies for skeletal muscle repair after atrophy. Our research suggests that there may be two potential pathways related to miR-133a in the mechanism of EA for disuse muscular atrophy (Figure 5). First, miR-133a may inhibit NLRP3 inflammasome activation to reduce muscle loss. Second, miR-133a regulates SIRT1 expression to induce skeletal muscle differentiation, which may help promote muscle tissue recovery.
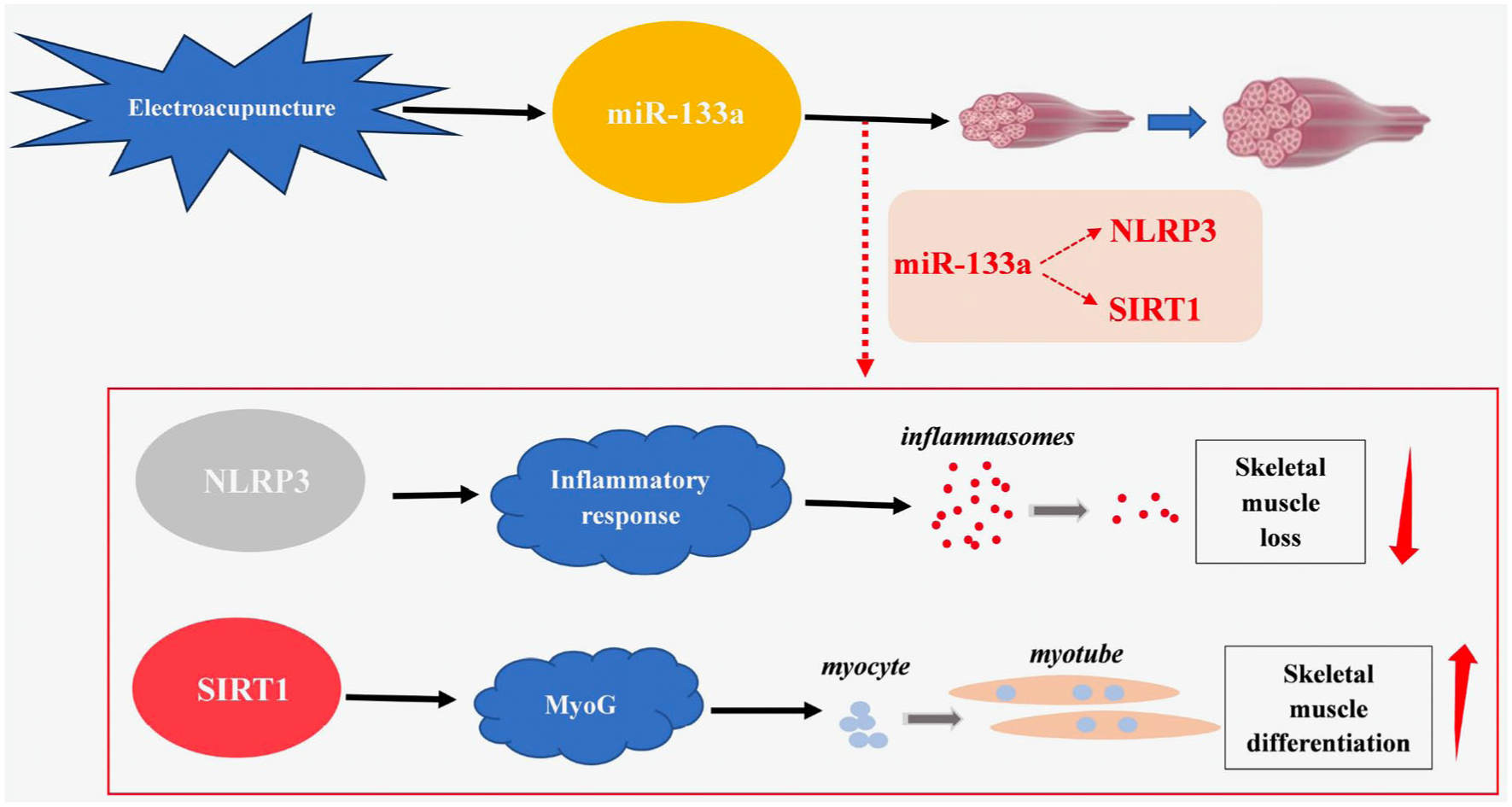
Electroacupuncture (EA) may accelerate the recovery of disuse muscular atrophy in two ways. First, activation of microRNA (miR)-133a may inhibit nucleotide-binding oligomerization domain, leucine-rich repeat (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome activation to decrease muscle loss. Second, activation of miR-133a may regulate Sirtuin1 (SIRT1) expression to facilitate skeletal muscle differentiation, and potentially promote muscle tissue recovery.
Footnotes
Acknowledgements
The authors would like to thank Army Military Medical University for providing the research platform for the experiment.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by the Space Medical Experiment Project of China Manned Space Program (grant no. HYZHXM 01017).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.