
Abstract
Background
Radiation-induced cognitive decline (RICD) is a common and debilitating late effect of cranial radiotherapy, particularly in long-term survivors of brain tumours and paediatric cancers. Despite advances in conformal radiation delivery and supportive care, progressive impairments in memory, attention and executive function continue to limit quality of life. Increasing evidence indicates that RICD is driven not by acute neuronal loss but by persistent neuroinflammation, oxidative stress, vascular dysfunction and metabolic failure within cognitive circuits. These converging processes highlight the need for disease-modifying targets that can regulate multiple pathological domains simultaneously.
Summary
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors that integrate inflammatory control, redox balance, lipid metabolism and mitochondrial function across neurons, glia and the neurovascular unit. Preclinical studies demonstrate that PPARγ and PPARα agonists, including pioglitazone and fenofibrate, prevent or attenuate cognitive decline after fractionated whole-brain irradiation even when structural injury persists. PPARβ/δ agonists suppress radiation-induced neuroinflammation, while emerging evidence suggests that ligands capable of stabilising white matter and glial phenotypes may further enhance cognitive resilience. In parallel, phytochemical PPAR modulators, dual-isoform ligands, and advanced delivery strategies expand the therapeutic landscape beyond first-generation metabolic drugs.
Key Message
PPARs represent an integrative, circuit-level target for modifying the delayed trajectory of radiation-induced brain injury. Strategic deployment of PPAR-directed therapies during and after cranial radiotherapy offers a biologically grounded and clinically actionable approach to preserving long-term cognitive function in brain tumour survivors.
Keywords
Radiation-induced Cognitive Decline: An Unmet Clinical Need
Primary brain tumours constitute a significant global health burden, particularly among children, where central nervous system (CNS) tumours are among the most common and morbid tumours in children and adolescent age groups (incidence ~3.9–4.0 per 100,000).1, 2 Cranial radiotherapy (RT) remains a cornerstone of treatment across tumour types because it effectively controls local disease and extends survival.3–6 Advances in systemic therapies and precision radiation delivery techniques have resulted in improved long-term survival. As the number of long-term survivors increases, there is a parallel rise in treatment-related sequelae, notably cognitive impairments that persist for years after completion of therapy, compromising the quality of life of affected individuals.7–9
Radiation-induced cognitive decline (RICD) manifests as progressive deficits in memory, attention, processing speed and executive function, undermining activities of daily living, employment and quality of life. RICD can affect 30% or more of patients at 4 months after cranial irradiation, rising to 50%–90% in those surviving beyond 6 months, with prominent impairments in learning and executive domains. Paediatric survivors display long-term declines that continue into adulthood, underlining the lifelong impact of cranial irradiation on neurocognitive development and function.9–11
Current strategies to mitigate RICD remain inadequate. The Food and Drug Administration-approved N-methyl-
Beyond symptomatic management, multiple signalling pathways have been implicated in the pathogenesis of RICD, including NMDA receptor-mediated excitotoxicity, renin–angiotensin–aldosterone system signalling, receptor for advanced glycation end products–high-mobility group box 1 (RAGE–HMGB1) axis, and mitochondrial and lipid metabolic dysregulation.10, 14 These pathways converge on chronic glial activation, oxidative injury and synaptic dysfunction, which drive progressive cognitive decline long after radiation exposure. The diversity of these targets highlights that RICD is a network disease rather than a single-pathway disorder, necessitating interventions capable of integrating immune, metabolic and neuronal control.
Peroxisome Proliferator-activated Receptors as Integrative Regulators of the Inflammatory–Metabolic Axis in Radiation-induced Cognitive Decline
As outlined above, RICD emerges from a persistent imbalance across three coupled biological domains: neuroinflammation, oxidative stress and mitochondrial–lipid metabolism, rather than from acute neuronal loss alone.10, 15–18 Peroxisome proliferator-activated receptors (PPARs) bridge these domains, making them well suited for therapeutic intervention in the irradiated brain. The structural organisation of PPARs and the core mechanisms underlying their ligand-dependent transcriptional regulation of metabolic and redox homeostasis are illustrated in Figure 1.
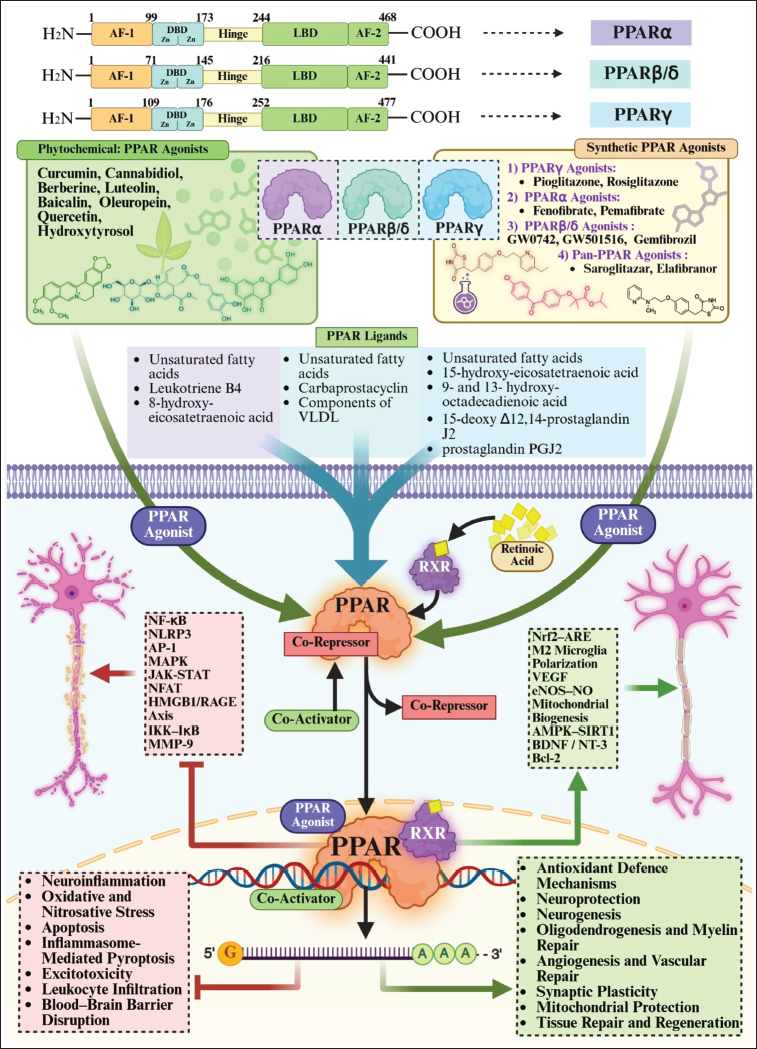
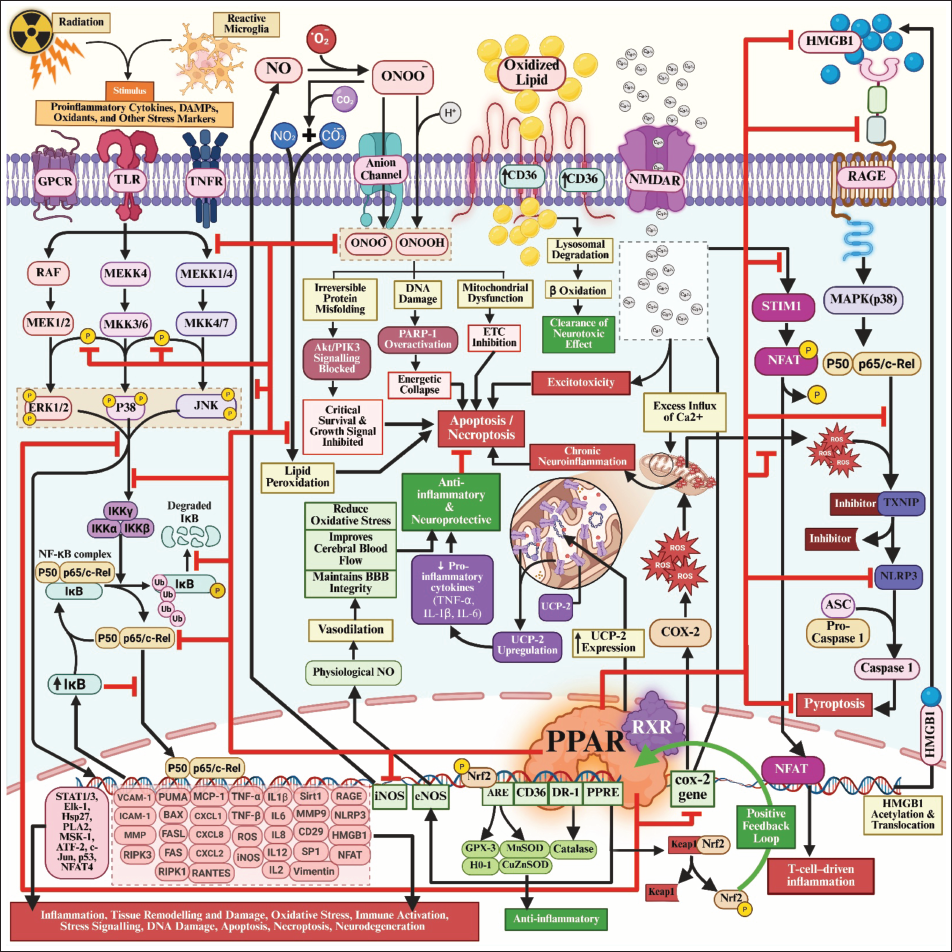
Following cranial irradiation, microglia and astrocytes enter a chronically activated state, releasing cytokines and danger-associated molecular patterns such as HMGB1, which engage RAGE and sustain nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-dependent inflammatory loops.14, 19–22 PPAR activation directly attenuates this feed-forward signalling through transrepression of NF-κB, signal transducer and activator of transcription (STAT) and AP-1, thereby reducing tumour necrosis factor-alpha (TNF-α), IL-1β, COX-2 and iNOS transcription.23–25 In parallel, PPARs enhance redox homeostasis via functional coupling to Nrf2, boosting antioxidant defences (HO-1, NQO1 and glutathione enzymes) that limit reactive oxygen species (ROS)-driven propagation of RAGE and inflammatory signalling.26–28
Crucially, PPARs reprogramme mitochondrial and lipid metabolism, restoring β-oxidation, adenosine triphosphate generation and membrane homeostasis needed for synaptic transmission and myelin maintenance.29–31 This metabolic support explains why PPAR agonists preserve cognition even when classical histological endpoints (e.g., neurogenesis or microglial counts) remain abnormal in RICD models. Because microglia, astrocytes, oligodendrocytes and neurons all express functional PPARs, receptor activation provides cell-type–spanning control over inflammatory homeostasis, oxidative stress and energetic capacity within hippocampal and frontal circuits that subserve memory and executive function. 32
Radiation simultaneously activates NF-κB, RAGE and ROS; PPARs suppress all three and restore metabolic competence.14, 25, 32 This network-level control distinguishes PPAR agonists from single-pathway inhibitors and positions them as circuit stabilisers capable of modifying the delayed trajectory of radiation injury. This systems-level interaction between inflammatory, oxidative and metabolic signalling networks and their modulation by PPARs is depicted in Figure 2.
The distinct CNS cell-type distribution, functional roles and pathway crosstalk of PPARα, PPARβ/δ and PPARγ that underpin their differential relevance to RICD are summarised in Table 1.
Peroxisome Proliferator-activated Receptor (PPAR) Isoforms, Central Nervous System (CNS) Relevance, Disease Linkage and Pathway Crosstalk.

Pharmacological Peroxisome Proliferator-activated Receptor Agonists in Radiation-induced Cognitive Decline
The preclinical evidence supporting PPAR agonists as neuroprotective agents in RICD derives principally from rodent models of fractionated brain irradiation and mechanistic studies in cultured cells. Across isoforms, PPAR activation has shown the capacity to mitigate the delayed and progressive cognitive impairments induced by cranial irradiation, but the strength of effect and mechanistic signatures differ by agonist class. This section reviews experimental outcomes for PPARγ, PPARα and PPARβ/δ agonists.
Peroxisome Proliferator-activated Receptor Gamma Agonists
Pioglitazone
The most direct evidence that PPARγ activation can prevent RICD comes from rodent fractionated whole-brain irradiation (fWBI) models. In a seminal study, early and continuous administration of the PPARγ agonist pioglitazone during cranial irradiation prevented subsequent deficits in hippocampal-dependent cognitive tasks compared to irradiated controls. 39 Adult male Fischer-344 rats received 40 or 45 Gy WBI delivered as two fractions per week over 4 or 4.5 weeks, mimicking delayed radiation injury rather than acute toxicity. Pioglitazone was administered in the diet (120 ppm) either before, during, and after WBI, or after completion of irradiation.
Rats exposed to WBI alone developed a significant decline in hippocampal-dependent cognitive performance. In contrast, animals that received pioglitazone before and during irradiation, whether continued for a longer duration (4–54 weeks) after irradiation, remained fully protected and performed equivalently to sham-irradiated controls. When pioglitazone was initiated only after WBI, cognitive performance showed a non-significant improvement but remained impaired.
Importantly, although pioglitazone robustly preserved cognitive performance, the study did not examine the underlying cellular or molecular mechanisms. No assessments of neurogenesis, microglial activation, cytokine signalling or oxidative injury were performed, leaving the biological basis of this protection unresolved. Notably, this remains the only study demonstrating that a PPARγ agonist can prevent RICD in a fWBI model. This lack of mechanistic follow-up represents a major knowledge gap, but also creates an opportunity: defining how pioglitazone confers cognitive resilience could enable the rational development of related PPARγ modulators, including rosiglitazone and selective next-generation agonists, for the radioprotection of the brain.
A phase I dose-escalation clinical trial demonstrated that pioglitazone was well tolerated in patients with primary and metastatic brain tumours receiving cranial radiotherapy, with no dose-limiting toxicities observed at daily doses up to 45 mg. 41 Pioglitazone was administered concurrently with radiotherapy and continued for 6 months post-treatment. Only isolated grade ≥3 adverse events were reported, and their attribution to pioglitazone was considered possible rather than definitive. These findings establish the safety and tolerability of pioglitazone in non-diabetic brain tumour patients undergoing radiotherapy and support its evaluation in future efficacy trials targeting RICD.
Pioglitazone also shows direct antitumour activity in glioma models, supporting its dual relevance for brain tumour patients. Oral or intracerebral pioglitazone reduced glioma growth, invasion and proliferation in xenograft models and achieved measurable brain concentrations, confirming blood–brain barrier (BBB) penetration. 42 In an orthotropic glioblastoma model, pioglitazone combined with statins reduced tumour volume and macrophage infiltration without added toxicity, indicating the potential to simultaneously protect cognition and limit tumour progression. 43 These findings indicate that pioglitazone may simultaneously protect normal brain tissue from radiation injury while limiting tumour progression, a highly desirable profile for RICD-directed therapy in brain cancer.
Rosiglitazone
Although rosiglitazone has not been evaluated directly in RICD models, multiple studies have shown that it targets the same neuroinflammatory and oxidative pathways that drive delayed radiation injury. In rats exposed to systemic lipopolysaccharide, rosiglitazone (1–5 mg/kg) reversed hippocampal-dependent learning and memory deficits while reducing amyloid-β accumulation, astrocyte activation and pro-inflammatory mediators (IL-6, TNF-α, nitric oxide and lipid peroxidation) and restoring antioxidant enzyme activity in hippocampal tissue. 44
At the cellular level, rosiglitazone suppresses macrophage and microglial inflammation through PPARγ-dependent inhibition of NF-κB signalling, as demonstrated by reduced p65 phosphorylation and cytokine release following inflammatory challenge. 45 In vivo, rosiglitazone also shifts activated microglia away from a pro-inflammatory phenotype and prevents neuronal loss in models of excitotoxic brain injury, demonstrating its capacity to modulate neuroglial injury states that resemble those induced by irradiation. 46 Importantly, rosiglitazone also inhibits inflammasome activation in radiation-exposed tissues, suppressing NLRP3, caspase-1, IL-1β and TNF-α in irradiated macrophages and rat intestine, a pathway increasingly implicated in radiation-induced neuroinflammation. 47
Together, these findings establish that rosiglitazone engages PPARγ-dependent inflammatory and oxidative injury pathways that closely parallel those driving RICD, providing a strong mechanistic rationale for its evaluation as a radioprotective cognitive modulator.
Peroxisome Proliferator-activated Receptor Alpha Agonists
Fenofibrate
Fenofibrate is one of the most comprehensively validated PPAR agonists in radiation-induced brain injury. Its neuroprotective effects have been demonstrated across both single-dose and fWBI models, with rigorous cellular and behavioural endpoints.
In a PPARα knockout study, wild-type and PPARα-knockout mice received 0.2% (w/w) dietary fenofibrate and a single 10-Gy dose of 137Cs γ-irradiation. 48 Whole-brain irradiation (WBI) significantly reduced hippocampal neurogenesis 2 months later, as assessed by BrdU/NeuN double labelling, and increased CD68-positive microglial activation. Fenofibrate prevented both neuronal loss and microglial activation in wild-type mice, but not in PPARα-knockout animals, establishing a PPARα-dependent neuroprotective mechanism.
In a more clinically relevant fractionated irradiation model, young adult Fischer-344 × Brown Norway rats received 40 Gy fWBI (two 5-Gy fractions per week for 4 weeks), with continuous dietary fenofibrate beginning 1 week before irradiation. 34 At 6–7 months post-irradiation, fWBI caused a significant deficit in perirhinal cortex-dependent novel object recognition, while hippocampal-dependent water maze performance remained intact despite marked reductions in neurogenesis and increased ED1-positive microglial activation. Fenofibrate fully prevented the perirhinal cortex–dependent cognitive deficit, yet did not normalise neurogenesis or microglial activation, demonstrating that PPARα activation preserves functional cognition despite persistent structural injury. This dissociation supports a model of circuit-level metabolic resilience rather than histological normalisation.
Fenofibrate also protects radiation-exposed vasculature, restoring redox balance, nitric oxide signalling, inflammatory responses, and endothelial integrity after irradiation. 49 As cerebrovascular dysfunction contributes to delayed cognitive decline, this endothelial protection likely synergises with hippocampal and cortical effects.
Beyond neuroprotection, fenofibrate exhibits tumour-selective radiomodulatory activity. In glioblastoma models, fenofibrate (50 µM) radiosensitised U87 cells by inducing oxidative stress, deoxyribonucleic acid (DNA) double-strand breaks and mitochondrial dysfunction, while protecting LN18 cells via lipid droplet-mediated drug sequestration and membrane stabilisation. 50 Modulating intracellular lipid trafficking converted fenofibrate into a tumour-selective cytotoxic radiosensitiser, demonstrating that tumour lipid metabolism governs its radiation response.
Together, these data identify fenofibrate as a PPARα-dependent neuroprotective agent that preserves cognition in the normal brain while selectively sensitising tumour cells to radiation, making it highly attractive for translational development in brain tumour patients to prevent RICD.
Pemafibrate
Pemafibrate is a next-generation, selective PPARα modulator and an exploratory candidate for RICD. It suppresses microglial activation through direct PPARα-dependent inhibition of NF-κB and cytokine expression, distinguishing it mechanistically from fenofibrate, which also engages SIRT1 signalling. 51 Beyond immunomodulation, pemafibrate prevents excitotoxic neuronal death by inhibiting phosphorylated c-Jun–mediated apoptosis. 52 In the hippocampus, pemafibrate restores PPARα–brain-derived neurotrophic factor (BDNF) signalling and neurogenesis and reverses stress-induced behavioural and cellular deficits, effects abolished by PPARα or BDNF inhibition. 53 These combined actions—microglial suppression, anti-apoptotic activity, and support of hippocampal neuroplasticity—support pemafibrate as a strong candidate for mitigating the inflammatory and neurogenic components of RICD.
Peroxisome Proliferator-activated Receptor Beta/Delta Agonists
GW0742
The PPARβ/δ agonist GW0742 was evaluated in mice following single-dose 10 Gy WBI. 37 Dietary GW0742 robustly suppressed radiation-induced neuroinflammation, blocking the early increase in IL-1β and extracellular signal-regulated kinase (ERK) activation, and reducing hippocampal microglial activation. However, it did not prevent early-delayed hippocampal-dependent cognitive impairment or restore hippocampal neurogenesis. The cognitive deficit detected at 3 months resolved by 6 months, indicating that the Barnes maze captured an early-delayed phase of injury rather than the progressive late-delayed cognitive decline observed clinically. Because GW0742 was not tested in perirhinal cortex-dependent paradigms that model late-phase RICD, its ability to modify clinically relevant cognitive decline remains unresolved.
Thus, GW0742 establishes that PPARβ/δ activation potently controls radiation-induced neuroinflammation, but whether this translates into durable cognitive protection remains an open question.
Gemfibrozil
Unlike GW0742, gemfibrozil engages PPARβ/δ to regulate both microglial activation and myelin gene expression. In human microglia, gemfibrozil suppressed inflammatory cytokines and CD11b expression through a PPARβ/δ-dependent mechanism. 54 In oligodendrocytes, gemfibrozil directly induced myelin gene transcription via PPARβ/δ recruitment to myelin gene promoters, an effect absent in PPARβ/δ-deficient cells. 55 Because chronic microglial activation and white-matter injury drive late-delayed RICD, this dual action positions gemfibrozil as a more promising PPARβ/δ ligand.
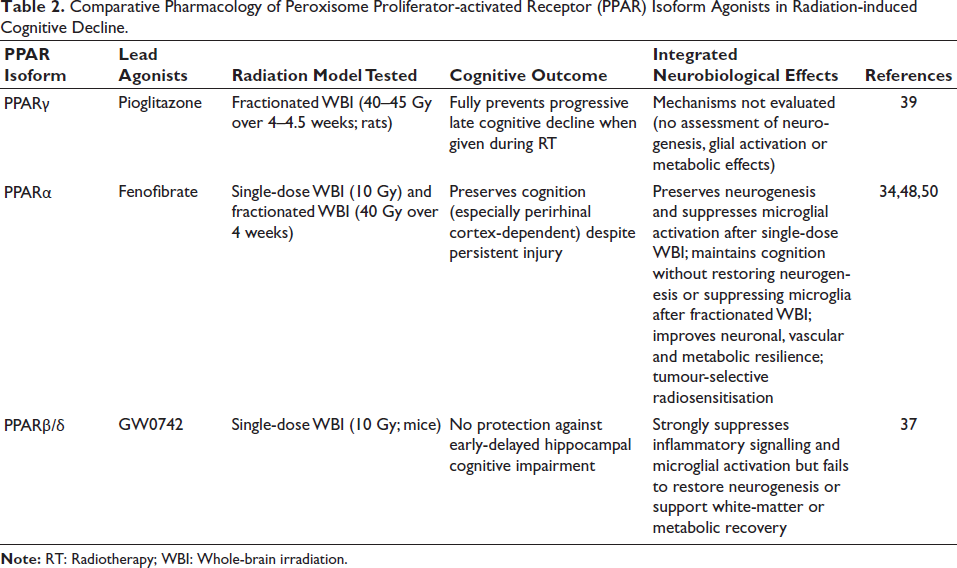
Overall, PPARβ/δ activation effectively suppresses radiation-induced neuroinflammation; however, its ability to prevent progressive late-delayed cognitive decline remains unclear, as existing studies have primarily used single-dose irradiation and early-phase hippocampal behavioural paradigms that do not fully recapitulate clinically relevant RICD. Given that progressive cognitive impairment is better modelled by fWBI and perirhinal cortex-dependent tasks, future studies should evaluate PPARβ/δ agonists under these translationally relevant conditions. Within this class, gemfibrozil warrants particular attention because it uniquely targets both neuroinflammation and white-matter integrity, two core biological drivers of RICD. A comparative overview of PPAR agonists across radiation models and cognitive outcomes is provided in Table 2.
Comparative Pharmacology of Peroxisome Proliferator-activated Receptor (PPAR) Isoform Agonists in Radiation-induced Cognitive Decline.
Limitations
Systemic Toxicity of Synthetic Peroxisome Proliferator-activated Receptor Agonists
Although synthetic PPAR agonists such as pioglitazone and fenofibrate provide compelling proof that PPAR signalling can modify RICD, these agents were developed for chronic metabolic diseases rather than long-term CNS use. PPARγ agonists are associated with weight gain, fluid retention and cardiovascular risk, whereas fibrates alter systemic lipid handling in ways that may be undesirable in paediatric patients and long-term cancer survivors.56, 57 Because RICD requires prolonged or prophylactic treatment, even modest systemic liabilities become clinically limiting. These safety constraints restrict dose escalation, duration of therapy, and use in vulnerable populations, reinforcing the need for brain-focused, isoform-balanced and pharmacokinetically optimised PPAR modulation rather than continuous systemic exposure.
Blood–Brain Barrier Permeability and Central Nervous System Exposure
Most PPAR agonists used in RICD models exhibit behavioural or histological effects consistent with CNS activity, yet quantitative intrabrain exposure remains largely undefined. Radiation alters BBB permeability, endothelial transport, lipid trafficking and efflux transporter activity, all of which influence drug distribution within hippocampal, cortical and vascular compartments.58, 59 As a result, systemic dosing does not reliably predict cell-type-specific target engagement in microglia, astrocytes, oligodendrocytes or neurons. Without rigorous neuropharmacokinetic and biodistribution data, it remains unclear whether cognitive benefit reflects direct neural PPAR activation or indirect systemic effects, limiting rational optimisation of dosing, timing, and compound selection.
Limitations of Preclinical Models of Radiation-induced Cognitive Decline in Assessing Peroxisome Proliferator-activated Receptor Agonists
Radiation Biology Mismatch
Most preclinical PPAR studies rely on single-dose irradiation, whereas patients receive fractionated, conformal X-ray or proton therapy. Dose, dose rate, fractionation and radiation quality fundamentally alter microglial activation, vascular injury, oxidative stress and metabolic responses, which in turn influence PPAR signalling.60–62 Models that do not recapitulate these parameters risk misrepresenting both injury mechanisms and therapeutic windows.
Age Mismatch
RICD is most clinically devastating in paediatric survivors, yet most studies use adult rodents. Neurogenesis, myelination, metabolic plasticity and PPAR-dependent transcription differ profoundly between the developing and mature brain, limiting extrapolation to childhood cancer populations.63, 64
Sex Bias
Nearly all RICD–PPAR studies use male animals, despite strong evidence that microglial activation, mitochondrial stress responses, and PPAR signalling exhibit sexual dimorphism.65, 66 This omission restricts translational relevance and obscures potential sex-specific therapeutic windows.
Behavioural Paradigm Mismatch
Many studies rely on a single hippocampal tasks (Barnes maze or Morris water maze), which capture early injury but incompletely models progressive, multimodal cognitive decline. Clinically relevant RICD affects memory, attention, executive function and social behaviour. A translational battery should integrate novel object recognition, Y-maze alternation, fear conditioning, social interaction and executive function assays to capture circuit-level dysfunction across hippocampal, cortical and striatal domains.67–69
Lack of Tumour–Radiation Interaction Studies
Few PPAR agonists have been evaluated in irradiated, tumour-bearing brain models, despite the need to ensure neuroprotection without tumour protection. Because PPAR signalling regulates lipid metabolism, oxidative stress and immune responses in cancer cells, rigorous testing in orthotopic glioma and paediatric tumour models is essential before clinical translation.
Narrow Exploration of the Peroxisome Proliferator-activated Receptor Ligand Space
Many PPAR agonists with strong activity in neurodegeneration, multiple sclerosis, traumatic brain injury and stroke have never been evaluated in RICD.70–75 Current knowledge relies on a small subset of repurposed metabolic drugs, leaving substantial untapped therapeutic space unexplored.
Absence of Central Nervous System Pharmacodynamic Biomarkers
Most studies lack direct evidence of which PPAR isoform is activated, in which cell types, and to what degree in the irradiated brain. Without validated CNS biomarkers of PPAR engagement, it remains difficult to distinguish target-driven neuroprotection from non-specific systemic effects, hindering dose optimisation and patient stratification.
Emerging Strategies
Phytochemical Peroxisome Proliferator-activated Receptor Modulators
The limitations of synthetic PPAR agonists have accelerated interest in phytochemical PPAR modulators as safer, long-term strategies for RICD. Many plant-derived small molecules act as partial or selective PPAR agonists, biasing receptor conformation towards anti-inflammatory and neuroprotective transcriptional programmes without triggering the adipogenic or cardiovascular effects associated with full synthetic agonists.76–78 Importantly, several phytochemicals exhibit robust BBB permeability and accumulate within microglia, astrocytes and neurons, enabling direct modulation of irradiated neural circuits.79, 80 The most translationally relevant phytochemical PPAR modulators—selected based on BBB penetration, PPAR engagement and anti-neuroinflammatory efficacy—are summarised in Table 3.
Phytochemical Peroxisome Proliferator-activated Receptor (PPAR) Modulators.
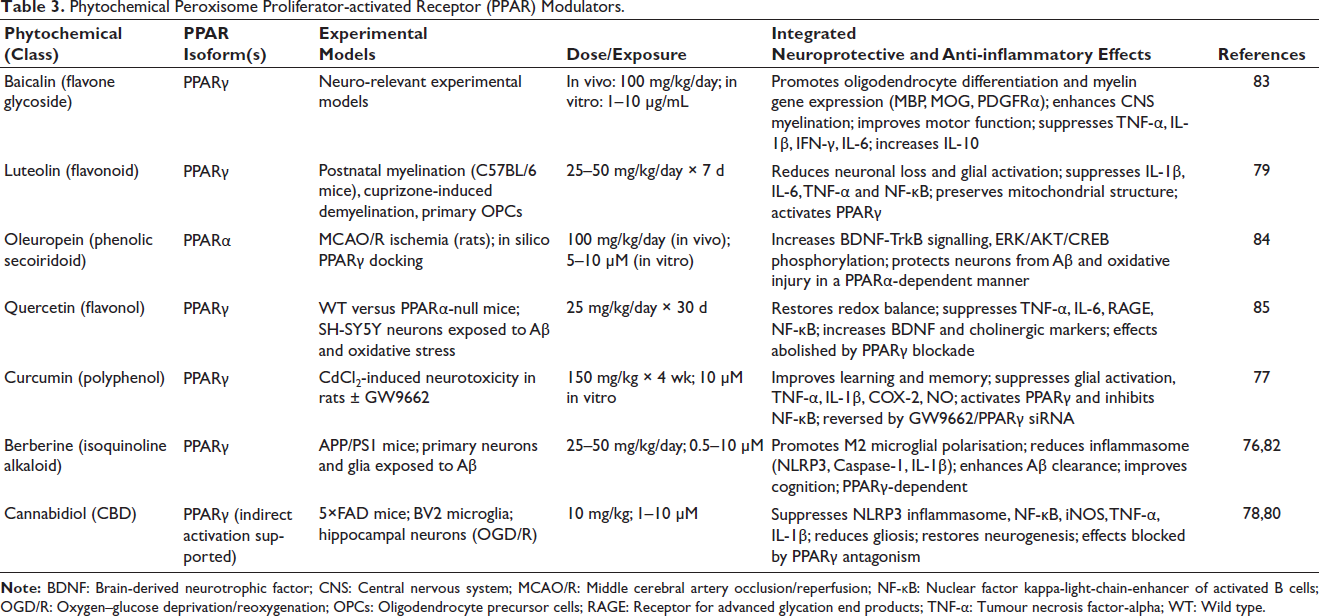
Unlike single-target drugs, phytochemical PPAR modulators also engage redox control, mitochondrial homeostasis and inflammasome regulation, providing network-level stabilisation of the irradiated brain.81, 82 Compounds such as curcumin, cannabidiol, berberine, luteolin, baicalin and oleuropein suppress microglial activation, restore mitochondrial function, and preserve synaptic integrity, often through PPAR-dependent mechanisms.26,77,79,80,82–84 Their favourable safety profiles and suitability for chronic oral administration make them particularly attractive for prophylaxis against late-delayed RICD.
Combination Strategies
PPAR signalling operates within a broader stress-response and metabolic network, making combination therapy a rational strategy for RICD. Pathological kinase pathways, such as CDK5-mediated repression of PPARγ, limit receptor responsiveness in injured neurons; suppressing these stress signals restores PPAR transcriptional activity and enhances neuroprotection. 86 Conventional anti-inflammatory agents also converge on PPAR-dependent transcriptional pathways, creating opportunities for synergistic pharmacology.87–89
Metabolic and redox vulnerabilities provide additional leverage. PPARα-driven GPX4 induction limits lipid peroxidation and ferroptotic stress, while coordinated PPARγ–PGC-1α activation enhances mitochondrial biogenesis and suppresses inflammation at lower drug doses.90, 91 Such combinations are particularly relevant to irradiated brain tissue, where oxidative damage and mitochondrial failure amplify neuroinflammatory injury.
Nanotechnology-enabled Delivery
A major obstacle in CNS PPAR therapy is subtherapeutic brain exposure. Many synthetic and phytochemical PPAR modulators suffer from poor solubility, rapid clearance and restricted BBB penetration.92, 93 Nanotechnology-based delivery systems overcome these limitations by enabling BBB transport, sustained parenchymal release and cell-selective accumulation.
Polymeric and lipid nanoparticles significantly increase brain concentrations of PPAR agonists, allowing therapeutic effects at lower systemic doses.94, 95 Pioglitazone-loaded nanoparticles enhance cognitive benefit and reduce neuroinflammation compared with the free drug, while dendrimer-based carriers preferentially target activated microglia. 94 Similar platforms improve CNS delivery of phytochemical PPAR modulators, such as curcumin and resveratrol, enabling brain-selective PPAR engagement with reduced systemic toxicity.96, 97
In Silico and Artificial Intelligence-guided Discovery Pipelines
Artificial intelligence and computational modelling are transforming PPAR drug discovery for complex disorders such as RICD. Network-level modelling shows that balanced PPARα/γ activation optimally restores inflammatory homeostasis, whereas unopposed activation drives maladaptive immune extremes. 98 Machine-learning models now predict PPAR–ligand affinity and isoform selectivity, enabling rapid prioritisation of large chemical libraries. 99
Structure-guided pipelines integrating docking, molecular dynamics and free-energy calculations support the design of partial and biased agonists that retain neuroprotective transcriptional programmes while minimising adverse effects.100–102 These approaches are especially powerful for identifying brain-penetrant, isoform-balanced PPAR modulators optimised for chronic CNS use.
Translational Biomarkers
Effective clinical translation requires biomarkers that reflect central PPAR engagement rather than peripheral metabolism. Microglial immunometabolic states, lipid handling and mitochondrial function provide biologically grounded readouts of PPAR activity.103, 104 Nuclear-receptor crosstalk, including PPAR–liver X receptor–apolipoprotein E signalling, further shapes lipid-dependent inflammatory phenotypes. 105
Imaging modalities such as translocator protein positron emission tomography provide non-invasive measures of microglial activation that correlate with cognitive decline and therapeutic response.106, 107 Together, molecular, metabolic and imaging biomarkers create a coherent framework for monitoring PPAR-targeted interventions and stratifying patients most likely to benefit.
Summary
RICD remains one of the most clinically challenging late effects of cranial radiotherapy, with limited options for prevention or disease modification. By coordinating inflammatory regulation, oxidative stress control, and cellular energy metabolism across neurons, glia and the neurovascular unit, PPARs occupy a central position in the biology of delayed radiation injury. Evidence from fractionated irradiation models demonstrates that PPARγ and PPARα agonists preserve cognitive function even when structural injury persists, while emerging data suggest that PPARβ/δ ligands capable of stabilising microglial activation and white-matter integrity may further broaden therapeutic potential.
Unlike single-pathway anti-inflammatory approaches, PPAR modulation supports circuit-level resilience by integrating immunometabolic and redox control within vulnerable cognitive networks. The expanding availability of repurposed drugs, selective modulators, dual-isoform ligands and brain-permeable phytochemicals provides a growing toolkit to exploit this biology in a translational context. Together, these advances position PPAR-directed strategies as a promising framework for mitigating long-term cognitive injury in survivors of cranial radiotherapy.
Footnotes
Acknowledgement
IKP is recipient of ICMR-Emeritus Scientist grant (No. HRD/IES-2025/4, dated 08.04.2025).
Authors’ Contribution
All authors contributed to the conception and design of the review, development of the analytical framework, literature curation, and synthesis of the data. All authors reviewed, edited and approved the final manuscript and agree with its content and submission.
Declaration of Conflict of Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Patient Consent
No patient consent was necessary for this review since it solely involved analysis of data from previously published sources.
Statement of Ethics
Ethical approval was not required for this review as the study was based exclusively on previously published literature.