
Abstract
Cerebellar ataxia, mental retardation, and dysequilibrium syndrome 4 (CAMRQ4) is a heterogenous group of genetic disorders that have been grouped based on their shared clinical features. CAMRQ4 should be suspected in patients presenting with ataxia, mental retardation, hypotonia, microcephaly, choreoathetoid movements or chorea, ophthalmoplegia, and global developmental delay, even if brain magnetic resonance imaging appears to be normal. We describe a family for whom reverse phenotyping played a crucial role in achieving the correct diagnosis of CAMRQ4 in the index child and prenatal diagnosis for the subsequent pregnancy. We describe the multidisciplinary approach that was followed during the pandemic, ensuring that the family was appropriately guided and informed.
Keywords
Introduction
Cerebellar ataxia, mental retardation, and dysequilibrium syndrome 4 (CAMRQ4) is a heterogenous group of genetic disorders that have been grouped based on their shared clinical features. 1 Four variants of the syndrome (type 1-4) have been identified thus far, and each one differs in terms of clinical features and genetics. Four genes have been reported to be responsible for this genetically heterogenous disorder: VLDLR (type 1), WDR81(type 2), CA8 (type 3), and ATP8A2 (type 4). 1
ATP8A2 gene mutation has emerged as a cause of novel neurological phenotype characterized by global developmental delays, severe hypotonia, and hyperkinetic movement disorder, the latter being important distinguishing feature. Optic atrophy is common and may only become apparent in the first few years of life, necessitating repeat ophthalmologic evaluation in older children. 2 In humans, ATP8A2 is mainly expressed in brain tissue, with highest levels in cerebellum as well as in retina and testis. Cerebellum is a crucial regulatory organ for motor coordination and this expression pattern is consistent with CAMRQ. 3 CAMRQ4 should be suspected in patients presenting with ataxia, mental retardation, hypotonia, microcephaly, choreoathetoid movements or chorea, ophthalmoplegia, and global developmental delay, even if brain magnetic resonance imaging (MRI) appears to be normal.
Here, we describe a family for whom reverse phenotyping played a crucial role in achieving the correct diagnosis of CAMRQ in the index child and prenatal diagnosis for the subsequent pregnancy. Reverse phenotyping has changed the usual diagnostic algorithm by making it possible to undertake phenotyping directed by genomic testing. 4 We also use this case to demonstrate the multidisciplinary approach that was followed during a pandemic, ensuring that the family was appropriately guided and informed. The SARS-CoV-2 outbreak and the consequent nationwide lockdown had significantly disrupted the delivery of health-care services, 5 including genetic testing and counseling. To ensure continuity of genetic counseling at a large university hospital, our team transitioned to fully remote telephone genetic counseling and testing services. However, the transition was not smooth in all domains of genetic counselling; prenatal diagnosis posed a number of challenges especially in cases where the re-evaluation of the index family after genetic testing needed to be performed before prenatal testing could be offered, as demonstrated in the following case report.
Case Report
The index child was a 6-year-old male, born by term vaginal delivery, with birth weight of 2.6 kg. There was no perinatal asphyxia but the child had global development delay floppiness, no head control, and was bed bound. He could recognize parents and occasionally say monosyllables. The MRI was normal. In the year 2016, multiplex ligation-dependent probe amplification of the SMN1 gene was performed and showed no deletion of exon 7 and 8. At that point, the parents were advised to do the next generation sequencing (NGS)-based clinical exome sequencing (CES), but they deferred.
In the year 2020, the mother (24-years old, G3P1L1A1) was referred for first trimester scan at 12 weeks. The fetus was structurally normal, nuchal translucency scan was within normal limits, and the first trimester screening reported a low risk for chromosome aneuploidies. The couple had a third degree consanguineous marriage and no significant familial or medical history was documented (Figure 1). The couple had a teleconsultation and were counselled about the need for an expedited evaluation and testing of the index child to enable direct testing by amniocentesis at 16 to 17 weeks of gestational age. However, in view of the nationwide lockdown, the parents were unable to bring the child for a detailed clinical evaluation. Consequently, on obtaining consent from the couple, a home-based sampling for the child was organized by the perinatal team.
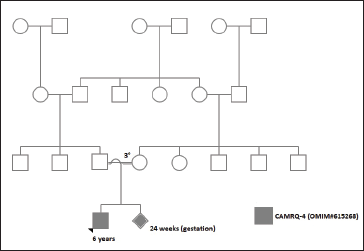
CES showed a deletion in exon 27 of ATP8A2 gene (CAMRQ-4: Online Mendelian Inheritance of Man [OMIM] #615268; ENST00000381655.7)3, 4 classified as a copy number variant of uncertain significance. The child was subsequently reviewed by a team consisting of a neurologist, ophthalmologist, and geneticist, and reverse phenotyping was carried out. While the MRI-brain was normal again, clinical and ophthalmic examination revealed choreoathetosis, global developmental delay, optic atrophy, and hypotonia, all of which were consistent with the CAMRQ syndrome. 5 With this background clinical information, the genetic report was reclassified as likely pathogenic using published variants in literature and a set of diseases databases which included ClinVar, OMIM, Genome-wide Association Study, and the Human Gene Mutation Database (HGMDv2019.4). Common variants were filtered based on allele frequency in 1000Genome Phase 3, gnomAD (v2.1), EVS, dbSNP (v151), 1000 Japanese Genome, and an internal Indian population database maintained by the service provider. The parents were counselled regarding the need for simultaneous parental testing and prenatal invasive testing due to the constraints of time.
Subsequently, amniocentesis was performed at 17 weeks 4 days and processed for a polymerase chain reaction (PCR) assay designed to detect the deletion in the exonic region 27 of the ATP8A2 gene. The PCR amplification was performed simultaneously on the fetus, index child, mother, father, and a healthy control to rule out the deletion. While the fetus and index child showed no amplification (homozygous deletion) for the exonic region 27 of the ATP8A2 gene, expected PCR bands were observed in the parents and healthy control. The heterozygous carrier state in the parents could not be confirmed with this method since a very large deletion was suspected. The couple was informed that the fetus was likely to be affected with the same disorder as their first child though the target scan was normal. The couple opted for termination of pregnancy.
Discussion
CAMRQ is a rare genetically heterogeneous cerebellar ataxia with mental retardation and dysarthric speech, with or without quadrupedal gait. Multiple consanguineous families have been reported with autosomal recessive inheritance of the condition, indicating that there is a 25% risk of recurrence if the parents are carriers.
Mutations in ATP8A2 were initially identified in a family with a clinical phenotype of CAMRQ syndrome. 3 ATP8A2 is highly expressed in the brain, spinal cord, retina, and testis. 6 The core phenotype associated with mutations in ATP8A2 gene is encephalopathy, intellectual disability, severe hypotonia, psychomotor delay, chorea, and optic atrophy without obvious radiographic evidence of cerebellar atrophy, whereas hearing loss appears to be a secondary phenotype. ATP8A2-associated disease and mutations within ATP8A2 should be considered when patients present clinically with chorea, vision impairment, severe neonatal hypotonia, and encephalopathy. 7 In the study by Alsahli and Alfadhel, 1 4 females and 1 male patient were included. They were all the offspring of consanguineous marriages, and each family harbored a different mutation affecting the ATP8A2 gene. Ataxia, mental retardation, microcephaly, hypotonia, and global developmental delay were evident in all the children. Choreoathetoid movement was present in 80% while ophthalmoplegia was present in 60%. All of children were on wheelchairs and none of them achieved ambulation. Their brain MRI and the biochemical laboratory investigations were normal. In the other study by McMillan et al, 2 despite the variability in mutation type and location, the patients in this series showed remarkable phenotypic similarity with all having profound cognitive impairment, severe and persistent hypotonia, and chorea or choreoathetosis. The identification of the latter feature is particularly important in distinguishing patients with ATP8A2 mutations from the many other genetic causes of cognitive impairment. Optic atrophy and, less commonly, ophthalmoplegia and ptosis can also be seen which may be helpful in establishing this diagnosis.
This case allowed us to document our experience in the shift to telehealth in response to the COVID-19 pandemic, in the context of reverse phenotyping, prenatal diagnosis, and genetic counseling. As these disorders usually have an early onset, genetic counseling is an important clinical tool for preventing new cases, especially for young couples with affected first child. Their risk of having an affected child in subsequent pregnancies is 25%. Prenatal diagnosis is proposed when the disease is well diagnosed and the causative mutation in the family is identified. 8
It has also demonstrated the power of reverse phenotyping in the interpretation of CES data directly in a clinical setting. We emphasize that interpretation of NGS data is an iterative process and its dynamic nature should be explained to families. For these reasons, reverse phenotyping of the index child and relatives, if necessary, should always be part of the diagnostic workup with a geneticist, neonatologist, pediatrician, obstetrician, and other specialties working closely to avoid misdiagnosis. Thus, our case highlights the importance of teamwork wherein the geneticist, clinical geneticist, perinatal team, and fetal medicine team collaborated in tandem to ensure that even in difficult circumstances like the pandemic, the family received appropriate advice and testing.
Footnotes
Acknowledgments
The authors are grateful to the family for their kind consent.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Statement
All procedures were in accordance with the ethical standards of the institutional committee on human experimentation and with the Helsinki Declaration. Informed consent was obtained in this study.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
The participant has consented to the submission of the article to the journal.