
Abstract
Terminal deletions of chromosome 6q are rare. The clinical findings are variable and most often include growth restriction, craniofacial anomalies, dysmorphism, microcephaly, cardiac anomalies, genitourinary anomalies, seizures, and tone abnormalities. Clinical manifestations are essentially due to differences in size and location of chromosomal deletions. This condition is usually associated with life-threatening morbidities often presented very early in neonatal period. Antenatal detailed ultrasound can help in identifying the condition which could then be confirmed by microarray after amniocentesis.
Here, we report a case of 6q deletion associated with facial, skeletal, and brain anomalies in a male preterm neonate. Our case shows many features that have hitherto not been observed in the reported cases worldwide. By reporting this case we intend to expand the already known database of information on the phenotypic features associated with 6q deletion syndrome.
Introduction
Deletions of the long arm of chromosome 6 are relatively rare. Deletions involve the loss of genes. Chromosomal rearrangements leading to 6q deletions are reported to be interstitial or terminal deletions. Origin of deletions could be de novo or could arise from structural chromosomal rearrangements like inversions. Specific chromosomal deletion syndromes are less likely to be suspected antenatally but may be incidentally discovered when chromosomal studies are done for other reasons or in view of abnormal antenatal USG scan findings. Postnatal diagnosis is suspected by clinical appearance and is confirmed by karyotyping or by other cytogenetic techniques such as chromosomal microarray analysis.
Case Report
One-day old baby weighing 2.175 kg was delivered, at 35+2 weeks, was one of the twins born to a 37-year old mother, primigravida, married nonconsanguineously for 2.5 years.
The current pregnancy was a twin intrauterine gestation conceived by in vitro fertilization with two embryo transfer (own sperm and ovum used). The cause of infertility was premature ovarian failure in mother with poor ovarian reserve. There was a history of two previous failed intrauterine conceptions before. An ultrasound was done at 20 weeks of gestation showed cardiac abnormality in form of mesocardia with cardiomegaly, enlarged stomach bubble, and cranial anomaly in form of dysgenesis of the corpus callosum and small cerebellum in the affected twin. As there were multiple abnormalities on antenatal USG, microarray was directly advised in anticipation of microdeletion that may go undetected by standard karyotype. The other twin showed normal ultrasound findings. Karyotype report of parents and that of the unaffected twin were normal.
Ultrasound in the affected twin done on follow-up at 33 weeks showed colpocephaly, agenesis of the corpus callosum, large stomach bubble. The antenatal Dopplers in the umbilical artery, showed increased resistance >95th centile, whereas the scan findings and the Dopplers of fetus B/ normal twin continued to be normal.
Antenatally betamethasone was given 2 doses 24 hours before delivery. The baby was born by LSCS delivery at 35+2 weeks in view of abnormal Doppler findings. APGAR score at 1 and 5 minutes were 3 and 5, respectively.
The baby was intubated in NICU/neonatal intensive care unit and was kept on a synchronous intermittent mode of ventilation. At 2 hours of life the neonate developed seizures that were intractable requiring phenobarbitone and levetiracetam.
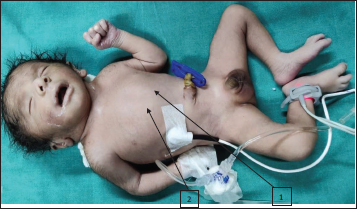
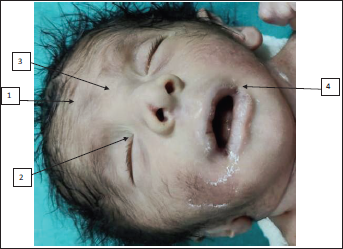
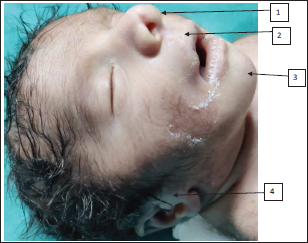
On examination, the baby had nonbulging open anterior fontanelle, bitemporal narrowing, mild hypertelorism, epicanthic folds, palpebral fissure short, low set ears with lobe attached, nasal bridge prominent, nose bulbous, philtrum smooth, macrostomia, high arched palate, retrognathia, wide-spaced nipples, and mild pectus carinatum (refer Figures 1-3). The limb dysmorphism comprised a Sydney line on the hands, arachnodactyly, and long narrow feet. There was severe kyphoscoliosis along with hypospadias, imperforate anus, and sacral pit. Testis was bilaterally descended with hypospadias. In anthropometry weight was 2.1 kg which is at 10th centile, head circumference was 30 cm which is below 10th centile, and length was 44 cm which is between 10th and 50th centile of Fenton growth chart.
Whole Body Image of the Baby: (1) Wide Spaced Nipple, (2) Pectus Carinatum.
(1) Narrow Forehead, (2) Epicanthal Folds, (3) Hypertelorism, (4) Wide Mouth Narrow Palpebral Fissure.
Baby Has (1) Bulbous Tip, (2) Smooth Philtrum, (3) Retrognathia (4) Low Set Ears Attached Lobule, Overfolded Helix.
USG cranium showed ventriculomegaly with dilatation of occipital and temporal horns bilaterally (Figure 4). Tiny grade I IVH was seen bilaterally, while an arching anterior cerebral artery with callosal artery was noted which ruled out complete agenesis of corpus callosum (Figure 5). However partial agenesis and thinning of callosum cannot be diagnosed on an ultrasound. Posterior fossa appeared prominent (prominent cisterna magna) and the vermis was visualized.
Dilatation of Bilateral Occipital and Temporal Horns.
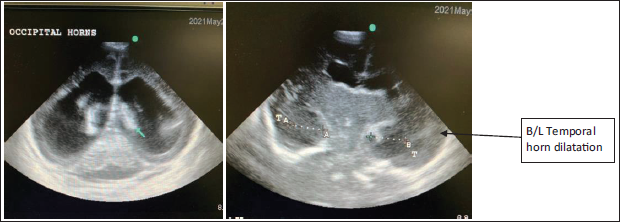
Ultrasound Picture with Arching Anterior Cerebral Artery with Callosal Artery Which Rules out Complete Agenesis of Corpus Callosum. However, Partial Agenesis and Thinning of Callosum Cannot Be Diagnosed on an Ultrasound.
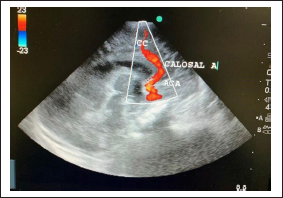
USG KUB/ultrasound kidney urinary bladder was normal. 2D ECHO/echocardiography showed evidence of cardiomegaly with poor cardiac contraction with dilated right ventricle with mild tricuspid regurgitation and patent ductus arteriosus of size 3 mm.
No other obvious structural cardiac malformation was noted. Although infantogram and MRI brain was planned, it could not be done as parents were not willing for any further investigations or treatment.
The baby had syndromic features with 6q deletion. Antenatal (315K resolution) microarray suggested 6q25.3-q27 microdeletion of 14.1 Mb size which contained about 125 genes. The microdeletion showed an overlap with the 6q deletion syndrome. For the microarray, Altum 750K CMA test was performed, which consists of 750k oligonucleotide probes across the genome including 550k unique nonpolymorphic probes, and 200k bi-allelic SNP (single nucleotide polymorphism) probes. The minimum resolution for detection is 200kb for losses, 400kb for gains and >5Mb for loss of heterozygosity.
The other twin was weighing 2.4 kg at birth and had normal vitals. Baby was accepting feeds well and was discharged home after three days of observation.
Poor prognosis was explained to the parents who then decided to withdraw the treatment after 24 hours of life.
Discussion
Deletion is a type of genetic aberration in which a part of the genetic segment is lost from the chromosome. Basically, 2 types of deletions are possible. Interstitial deletion, with the involvement of centromere, and the other being terminal deletion which does not involve the centromere. 1
Chromosome 6q deletion is a terminal deletion where a part of the long arm (q arm) of chromosome 6 is deleted from one of the copies. In most of the cases this deletion is not inherited and is likely to be a de novo occurrence, but they may rarely be transmitted by a normal parent who may carry a smaller microdeletion in the same region. 2 So, it is important to screen the parents by microarray or fluorescence in situ hybridization (FISH), as sometimes the microdeletion or microduplication that may be missed by microarray can be picked up by FISH.
Chromosome 6q24-q25 deletion syndrome is a very rare disorder with less than 15 publications and fewer than 50 reported cases so far in the world according to a recent literature review.
Pirola et al in 1998 first reported 6q deletion at 6q25 of about 8 cM, from D6S1496 to D6S437 in a 9-month-old girl who had hydrocephalus, muscular hypotonia, dolichocephaly, high-arched cleft palate, low-set ears, relatively long distal phalanges of fingers and agenesis of the corpus callosum on MRI. 3
Sukumar et al in 1999, reported a baby boy with a de novo interstitial deletion, chromosome 6 deletion (q25.1q26) with anomalies of the brain, genital organs, limbs, and feet. FISH using chromosome 6 painting probe ruled out an insertion. 4
Hopkin et al reported three cases, one of them was a male with a terminal deletion [del(6)(q25.2)] with hydrocephalus, retinal abnormalities, cleft palate, genital hypoplasia, and hydronephrosis. 5
Pen Shu et al in 2008 reported a 14.5 years old girl with 6q terminal deletion syndrome who had microcephaly, low frontal hairline, hypertelorism, depressed nasal bridge, bulbous nasal tip, large and low-set ears, micrognathia, high arched palate, short neck, with unusually short stature. To define the breakpoints, comparative genomic hybridization was done on the patient, which revealed that the aberrant chromosome 6 had a breakpoint at 6q25.3. 6
The latest case report was published in 2018 by Ritelli et al where he described a patient with congenital heart disease and dysmorphic features that included round face, wide forehead, almond-shaped eyes with ptosis, prominent columella, wide mouth with long flat philtrum, low-set ears, webbed neck, and pectus excavatum. Copy number variation analysis of exome had shown a deletion of chromosome 6q25.1 (chr6:149,056,337-151,113,208, GRCh37), comprising TAB2 gene. 7
We report a preterm twin delivery wherein one of the twins was diagnosed to have 6q25.3-q27 microdeletion of 14.1 Mb on chromosome 6. The exact genomic coordinates on microarray were 156447182_170605209 and the region encompassed 125 genes.
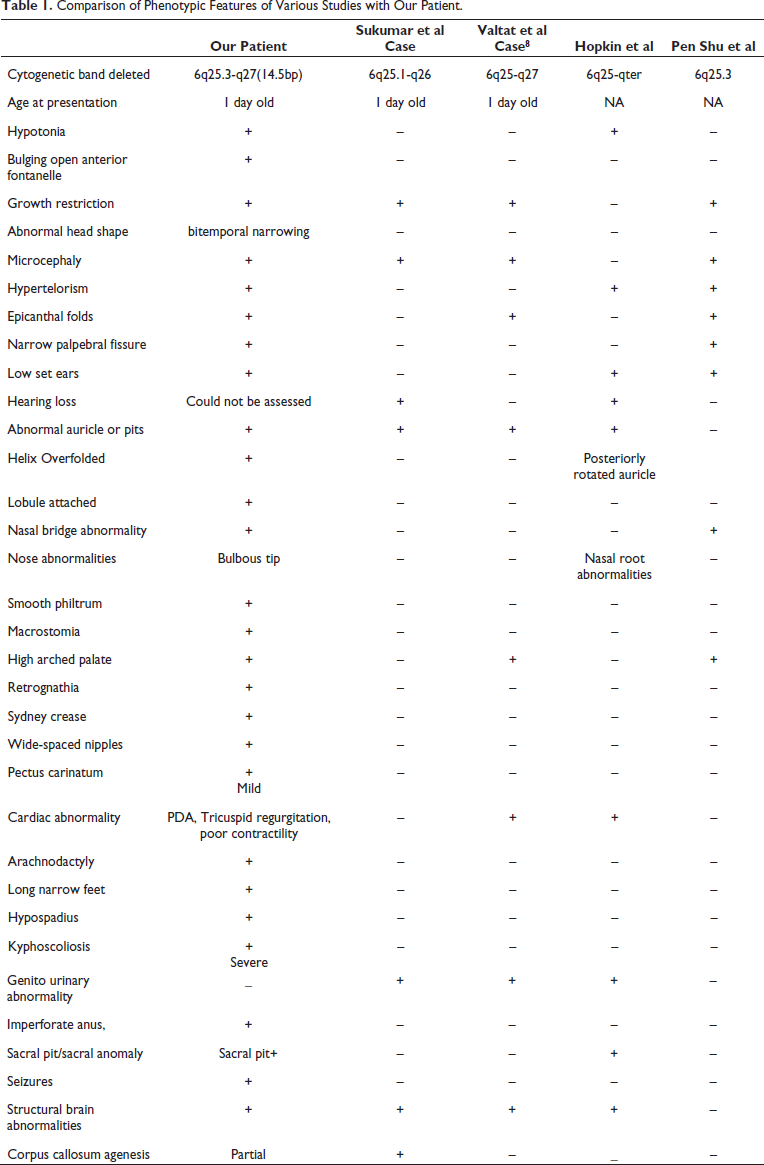
Our case shows many features that have hitherto not been observed in the reported cases worldwide (refer to Table 1 for comparison). By reporting this case we intend to expand the already known database of information on the phenotypic features associated with 6q deletion syndrome.
Comparison of Phenotypic Features of Various Studies with Our Patient.
We also want to emphasize the fact that early antenatal diagnosis with ultrasound and genetic analysis guides the clinician and the parents to decide on the future course of pregnancy. In view of grave prognosis and the lack of targeted treatment options, early antenatal diagnosis is of paramount importance and the clinician should have high index of suspicion so as to avoid missing this rare diagnosis.
Neonatologists are the first physicians to evaluate congenital anomalies or dysmorphism in babies and therefore need to be familiar with various physical differences in order to facilitate further screening for those occult malformations (refer to Figure 6).
Approach to a Dysmorphic Baby in NICU.
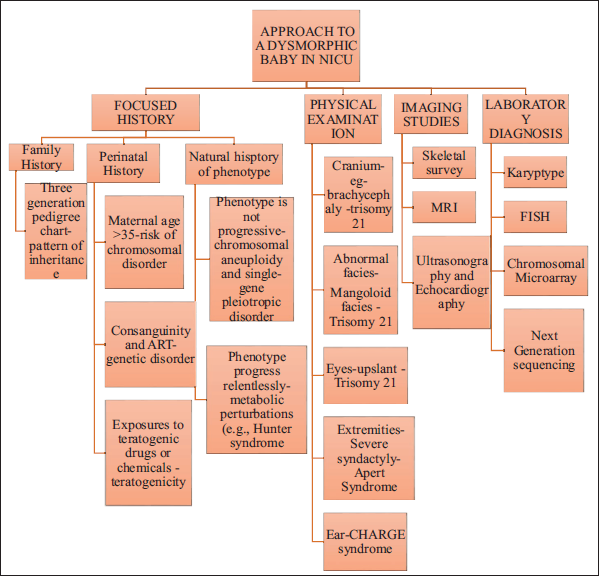
The history may provide some clues towards a possible genetic etiology. Therefore, consanguinity, history of dysmorphism in previous babies, previous miscarriages, age at conception, mode of conception (natural/in vitro fertilization), antenatal ultrasound scan findings, results of the prenatal markers (double/quadruple tests), and findings of invasive testing if carried out need to be reviewed properly. Intake of periconceptional multivitamins and folic acid or exposure to teratogenic drugs or agents must be documented. Detailed family history and pedigree drawing also give clues towards a possible genetic cause.
Along with the routine neonatal anthropometry, a proper head to toe examination must be carried out and findings must be noted down. These findings when collated together may point towards a minor or major dysmorphic syndrome. Internal organ evaluation by means of radiographs, ultrasound of abdomen/pelvis and brain, echocardiography, or an MRI brain in case of major neurological symptoms, etc, must be done as relevant.
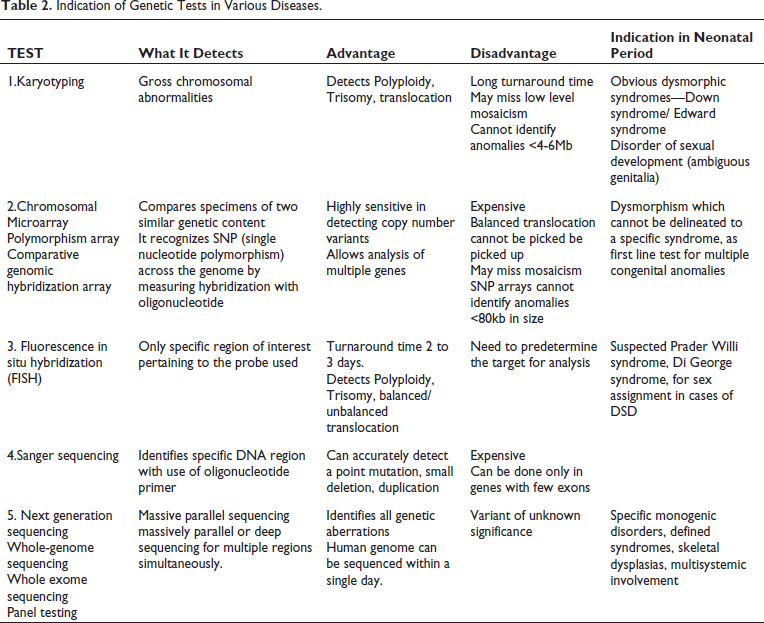
Very few syndromes such as Down syndrome, Patau syndrome, and Cornelia de Lange syndrome may have features typical enough to be diagnosed clinically at birth. The other syndromes may present with more subtle features which may evolve over time. Therefore, upon suspicion of a dysmorphic syndrome further work up must be initiated judiciously as suggested in Table 2.
Indication of Genetic Tests in Various Diseases.
Management of the dysmorphic neonates and genetic counselling are essential aspects of the approach to these patients. Benefits of early and accurate diagnosis help in providing anticipatory guidance and medical monitoring of patients for syndrome-specific medical risks that can help prolong and improve their quality of life. Once the diagnosis is made, the treating physicians can refer to published information on the natural history and management of particular syndromes through articles, genetics reference texts, online databases for more common disorders. The other major benefit of an accurate diagnosis is that it provides data for appropriate recurrence risk estimates for the parent’s future conception. Genetic evaluation and counselling by a clinical geneticist help in the correct interpretation of test results, gaining deeper insights into the condition and advising about preventive strategies to avoid recurrences in the family.
Authors’ Contribution
Anusha R, Srushti Nakade, and Chaitanya Datar were involved in the conceptualization of the manuscript, collecting patient data, conducting a literature search, and drafting the manuscript. All these three authors (Anusha Rao, Srushti Nakade, and Chetan Datar) have been designated as “First Authors” of this manuscript. Chaitanya Datar supervised the data collection, conducted a literature search, and revised the manuscript for the scientific content. Umesh Vaidya critically reviewed the manuscript for the scientific content. All 4 authors were involved in the clinical management of the patient and have approved the final manuscript submitted for publication. Chaitanya Datar will act as the guarantor and author for correspondence for the paper.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Statement of Informed Consent and Ethical Approval
Necessary ethical clearances and informed consent was received and obtained respectively before initiating the study from all participants.