
Abstract
Isovaleric acidemia (IVA) is an autosomal recessive inherited metabolic disease caused by the deficiency of isovaleryl-CoA dehydrogenase (IVD). The symptoms of IVA mimic sepsis, delaying the diagnosis. It can progress to coma and death if untreated. Thrombocytopenia and/or pancytopenia and hyperammonemia are the notable metabolic abnormalities. We report a neonate who presented to us with thrombocytopenia and encephalopathy. Tandem mass spectrometry was suggestive of IVA. Clinical exome sequencing identified a homozygous mutation in the IVD gene at codon 320 (p.Gln320His). The infant responded to specific treatments for IVA, such as carnitine and glycine supplementation. The infant attained appropriate development despite prolonged encephalopathy.
Introduction
Isovaleric acidemia (IVA) is a rare inborn error of metabolic disease caused by the deficiency of isovaleryl-CoA dehydrogenase (IVD). The prevalence of IVA ranges from 1 in 62,500 in Germany to 1 in 2,50,000 live births in the USA. 1 There are only a few cases reported from India.
The prognosis of IVA is better than for other organic acidemia. We report a neonate who presented to us after 11 days of birth with lethargy and poor feeding. He had severe encephalopathy, diagnosed with IVA based on tandem mass spectrometry (TMS), confirmed with clinical exome sequencing.
Case Report
A 12-day-old male infant presented to us with poor activity and refusal of feeds for the past 2 days. He was born to a primi mother via cesarean section for nonprogression of labor. There were no significant antenatal findings or history of inherited metabolic diseases in their family. The anthropometry measurement was as follows: weight 2.7 kg (10th percentile), length 45 cm (less than 3rd percentile), ponderal index of 2.96, and head circumference 32 cm (3rd percentile). The Apgar score was 8/10. The mother fed him predominantly breastfeeding and the term formula feed occasionally.
Physical examination at admission showed depressed sensorium, responded only to painful stimuli and intermittent jitteriness. There was mild erythema in the umbilical cord with pus discharge. The systemic examination was normal.
Investigation showed the leukocytes of 4700 cells mm−3, hemoglobin of 14.5 g dL−1, platelet count of 45,000 cells mm−3, and C-reactive protein (CRP) of 9 mg L−1. Serum calcium was 8.4 mg dL−1 and ionized calcium was 4 mg dL−1.
The differential diagnosis at that point was late-onset sepsis meningitis and congenital intrauterine infection.
Cerebrospinal fluid analysis showed 12 cells, neutrophil of 20%, lymphocyte of 80%, glucose of 81 mg dL−1, and protein of 43 mg dL−1. Cerebrospinal fluid herpes simplex virus polymerase chain reaction (CSF HSV PCR), urine cytomegalovirus PCR, and reverse transcription PCR (RT PCR) for COVID-19 were negative. For COVID-19, immunoglobulin G was positive, whereas immunoglobulin M was negative. Blood culture and CSF culture were sterile. Methicillin-sensitive staphylococcus was isolated from umbilical pus. The laboratory values are shown in Table 1.
Biochemistry Laboratory Values
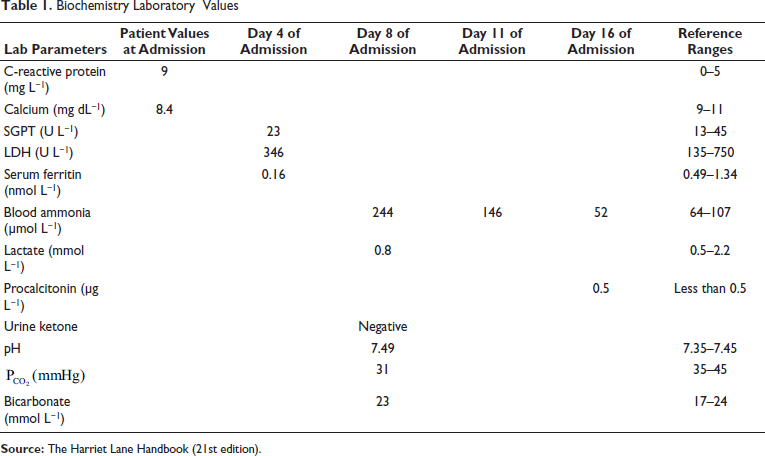
Magnetic resonance imaging was normal. He was evaluated for an inborn error of metabolism (IEM) as he had persistent thrombocytopenia and prolonged encephalopathy. We monitored encephalopathy using Thompson’s score. He had a score of 15/22 till the ninth day of admission. The platelet count remained less than 50,000 cells mm−3 for 18 days of admission. He developed pancytopenia on day 11 of admission (Hb 8 g dL−1, leukocyte count of 3000 cells mm−3, and platelet count 15,000 cells mm−3) and received five platelet transfusions and two times packed red blood cell transfusion. Blood ammonia on day 8 of admission was 416 µg dL−1 (244.26 µmol L−1), serum lactate was −7.5 mg dL−1 (0.83 mmol L−1), blood sugar was within normal limits, and urine ketone was negative. There was no specific urine odor to suggest a diagnosis of IEM.
He received the term formula feed through gavage feeding initially and kept nil per oral for 2 days when we identified hyperammonemia on day 8 of admission. After that acute empiric management of hyperammonemia started with parenteral vitamin b12 (Optineuron Injection) in maintenance intravenous (IV) fluid, protein-free diet, and drugs such as sodium benzoate, oral arginine, and IV carnitine.
TMS showed a high level of isovaleryl carnitine with decreased levels of free carnitine. Urine gas chromatography mass spectrometry (GCMS) shows an increased excretion of 3-hydroxyisovaleric acid and isovaleryl glycine.
The infant’s sensorium improved with a decline in the ammonia level. The platelet count improved after starting specific treatment for IVA, such as leucine-free diet and oral glycine on day 14 of admission. He developed thrombophlebitis on his left hand and subtle seizures in the form of conjugate deviation of eyes after 17 days of admission. Procalcitonin was positive (0.5 ng mL−1). He received IV antibiotics and antifungals. He accepted paladai feeding on day 20 of admission. He was discharged with IVA along with the prescription of following a low-protein diet (leucine-free diet, e.g., Pristine Balance Metanutrition), oral levetiracetam, carnitine, and glycine supplementation.
Clinical exome sequencing showed a homozygous missense variant in exon 9 of the IVD gene in chromosome 15 that resulted in the amino acid substitution of histidine for glutamine at codon 320 (p.Gln320His).
The infant received only special feed (leucine-free diet) because of the nonavailability of the mother’s milk till 6 months of age, followed by a low-protein diet. He was admitted twice for viral fever and gastroenteritis at 12 and 18 months of age and received IV fluids. He developed mild metabolic acidosis during admissions. The predominant symptom was vomiting that improved with hydration. He had axial hypotonia and squint noticed at 3 months of age. He received oral phenobarbitone for excess irritable cry at the age of 3 months. It was stopped after 2 months once irritability was reduced. He attended an intervention clinic regularly. He is now 2-and-a-half years old, and his development and neurological examination are appropriate for his age.
Discussion
IVA is an autosomal recessive disease caused by the deficiency of IVD. This enzyme is an intramitochondrial flavoenzyme that converts isovaleric acid to beta-methylcrotonyl-CoA, a step in a leucine catabolism pathway. 2
The clinical course of IVA infants is highly variable from the asymptomatic to severe form. The clinical phenotypes are of two types: neonatal, which is of the acute severe form, and the chronic intermittent form with onset in later infancy. The neonatal type accounts for more than half of the patients, and the survivors have frequent intermittent episodes during early infancy and decrease as they become old. 3 Recently a third group of individuals have been described who can be asymptomatic with mild biochemical abnormalities detected through newborn screening (NBS). 4
The clinical features of IVA are vomiting, tachypnea, lethargy, and stupor. The offensive sweaty feet odor is the characteristic feature of IVA. The metabolic abnormalities include severe acidosis, mild to moderate hyperammonemia, and hypocalcemia. Hyperammonemia in a neonate is seen in perinatal asphyxia, transient hyperammonemia of preterm, urea cycle disorder, organic acidemia, and fatty acid oxidation defect. If it is associated with ketoacidosis, it suggests organic aciduria; without acidosis, it suggests urea cycle disorder.
Thrombocytopenia, neutropenia, and/or pancytopenia are also common finding. 5 Additional clinical features include seizures, movement disorders (extrapyramidal movements and tremor), developmental delay, and intellectual disability. Pancreatitis, Fanconi syndrome, optic nerve atrophy, and cardiac arrythmia are rarely reported. 6
Our case had thrombocytopenia at admission and mildly elevated CRP, which makes us think of sepsis as the diagnosis. The prolonged encephalopathy and pancytopenia give a clue to workup for IEM. Blood gas showed pH of 7.49,
Sanjukta Dey et al. 7 reported a case of IVA in a neonate mimicking diabetic ketoacidosis and sepsis. Kelleher et al. 8 reported severe pancytopenia in two infants diagnosed with IVA, which could be because of arrest in the maturation of hematopoietic precursors by the accumulated toxic metabolites.
The diagnosis of IVA is established by the detection of accumulated derivatives such as isovaleric acid, three hydroxy isovaleric acid, and isovaleryl glycine by urine GCMS 9 and the detection of isovaleryl carnitine (C5 carnitine) in dried spots by TMS or identification of biallelic pathogenic variants in IVD by molecular genetic testing.
The treatment of acute metabolic crisis includes
Hydration Reversal of the catabolic state by giving glucose-insulin infusion and intralipid administration Correction of metabolic acidosis by sodium bicarbonate Treatment of hyperammonemia-N-carbamoylglutamate @ 100 mg kg−1 divided every 6 hours Facilitation of IVA excretion:
Long-term management includes a low-protein diet (safe limit of protein intake as prescribed by WHO/FAO/UNU 2007), carnitine, or glycine supplementation. Some centers use leucine-free amino acid mixture feed that is rarely needed in the management of IVA. 10 It has been found that the control of the endogenous protein breakdown is more effective than the restriction of dietary protein intake. 11
The special formula diet specific for IVA (Pristine Balance Metanutrition provides 15 g protein 100 g−1 powder) is available in India. 4 Metabolic dietician plays a vital role in long-term management, especially needed during the introduction of solid food.
Early diagnosis and treatment result in normal neurological outcomes, and the prognosis is good, as compared to other organic acidemia. 12 The inclusion of IVA in a universal NBS program is advisable as early diagnosis improves the outcome. 13
The risk of recurrence is 25% in every pregnancy as it is inherited autosomal recessively. The molecular diagnosis in this child helps in knowing the disease-affected status in future pregnancies.
Conclusion
The acute crisis of IVA manifests as severe vomiting and encephalopathy, mimicking sepsis; hence, one should also consider IEM in neonates having pancytopenia and prolonged encephalopathy. Early identification and appropriate treatment lead to normal neurological outcomes. Late diagnosis leads to neurological impairment; hence, the author recommends the inclusion of IVA in NBS for neonates at risk of IEM.
Footnotes
Acknowledgments
I thank Dr Raeshmi Ramalingam, MD pediatrics, and Dr Sasi Anand, DNB pediatrics, of Ramalingam Hospital for assisting in patient diagnosis and management.
Declaration of Conflicting Interest
The authors declare no potential conflicts of interest concerning this article’s research, authorship, or publication.
Ethical Approval
Ethical permission was not applicable for this article, as this is a review article drafted from various research articles and not from patients directly.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
The participant has consented to the submission of the article to the journal.