
Abstract
Background:
Familial Mediterranean fever (FMF) is a monogenic autoinflammatory disorder characterised by recurrent febrile attacks and serosal inflammation. Although Mediterranean fever gene (MEFV) gene mutations are central to disease pathogenesis, considerable phenotypic heterogeneity exists and the contribution of autoimmune comorbidities to disease expression remains insufficiently characterised in adult populations.
Objective:
This study aimed to evaluate genotype-phenotype associations and to assess the frequency and clinical impact of autoimmune comorbidities in a large cohort of adult patients with FMF.
Methods:
We retrospectively analysed 786 adult FMF patients diagnosed according to the Tel-Hashomer criteria. Demographic characteristics, MEFV mutation profiles, clinical manifestations during attacks, acute-phase reactants, treatment regimens and response to colchicine were systematically recorded. Genetic analyses were performed using sequencing-based methods. Statistical analyses were conducted using Statistical Package for the Social Sciences software.
Results:
Arthralgia was the most frequent clinical manifestation (89.8%), followed by abdominal pain and myalgia. Homozygous MEFV mutations, particularly M694V homozygosity, were significantly associated with increased disease severity, higher attack frequency and colchicine resistance. Autoimmune comorbidities were identified in 30.4% of patients, with ankylosing spondylitis being the most common (19.8%). Patients with homozygous M694V mutations exhibited higher rates of colchicine resistance (P = .021) and required interleukin-1 inhibitors more frequently (P = .003).
Conclusion:
FMF exhibits marked clinical heterogeneity influenced by both genetic background and coexisting autoimmune conditions. Homozygous M694V mutations are associated with severe disease and treatment resistance. Routine evaluation for autoimmune comorbidities may facilitate individualised treatment strategies in adult FMF patients.
Keywords
Introduction
Hereditary recurrent fevers represent a heterogeneous group of systemic autoinflammatory disorders, most of which are caused by monogenic mutations. Among these, familial Mediterranean fever (FMF) is the most prevalent condition. 1 FMF is an autosomal recessive inflammatory disease characterised by short, recurrent episodes of fever and serositis. The diagnosis is primarily clinical and often based on the widely accepted Tel-Hashomer criteria.2,3
FMF is particularly common in Mediterranean and Middle Eastern populations, with a notably high prevalence among individuals of Armenian descent.1,4 The causative gene, Mediterranean fever gene (MEFV), was identified approximately three decades ago and encodes pyrin, a 781-amino-acid protein involved in inflammasome formation. Upon activation by pathogen-associated or damage-associated molecular patterns, the pyrin inflammasome triggers the release of proinflammatory cytokines such as IL-1β and IL-18.5–7
Based on MEFV mutation status, patients are classified into homozygous, heterozygous and compound heterozygous genotypes, each potentially associated with distinct clinical phenotypes.5–7 Disease severity and complications, such as early onset, increased joint and organ involvement, sacroiliitis and amyloidosis, are more commonly observed in patients with homozygous mutations.8,9 During attacks, acute-phase reactants including white blood cell count (WBC), C-reactive protein (CRP), erythrocyte sedimentation rate (ESR) and fibrinogen are typically elevated.10,11
While colchicine remains the mainstay of FMF treatment and is effective in preventing attacks and amyloidosis, some patients experience persistent disease activity despite optimal dosing and are considered colchicine-resistant.12–14 In such cases, adjunctive therapies may be required.
FMF often coexists with other inflammatory rheumatic diseases, such as ankylosing spondylitis (AS), rheumatoid arthritis (RA), Behçet’s disease and vasculitis. 15 Although FMF is a well-described condition, clinical presentations and treatment responses can vary significantly between patients.
In addition to its classical autoinflammatory features, increasing evidence suggests that FMF may coexist with a variety of immune-mediated inflammatory diseases. Previous reports have described associations between FMF and conditions such as AS, Behçet’s disease, RA and psoriasis. These overlaps may reflect shared inflammatory pathways, genetic predisposition or environmental triggers that influence immune regulation. Understanding the frequency and clinical implications of such comorbidities is important, as overlapping conditions may alter disease presentation, complicate diagnosis and influence therapeutic decision-making.
This study retrospectively analysed a large cohort of 786 adult FMF patients to evaluate demographic characteristics, genotype distributions, clinical manifestations and comorbid autoimmune conditions. The influence of genetic variability and coexisting autoimmune diseases on disease severity, attack frequency and treatment outcomes was assessed. Additionally, the frequency and clinical impact of comorbid autoimmune disorders on the FMF disease course were examined.
Materials and Methods
This retrospective cohort study included adult patients diagnosed with FMF who were followed at a tertiary referral rheumatology centre between January 2010 and December 2020. FMF diagnosis was established according to the Tel-Hashomer criteria. Only patients aged ≥18 years with complete clinical, genetic and laboratory data were included in the analysis. Patients with insufficient medical records or uncertain FMF diagnosis were excluded.
Demographic data, including age, sex, number of siblings and family history of FMF, were recorded. Clinical characteristics comprised age at symptom onset, age at diagnosis, disease duration, frequency and type of FMF attacks and treatment history. The presence of comorbid autoimmune or chronic inflammatory diseases was systematically assessed based on clinical diagnoses documented in patient records and relevant laboratory or imaging findings. Autoimmune comorbidities included, but were not limited to, AS, Behçet’s disease, systemic lupus erythematosus and psoriasis.
Patients were identified through the hospital electronic medical record system using the diagnostic codes corresponding to FMF. All available clinical records were reviewed manually to confirm the diagnosis according to established clinical criteria. Demographic characteristics, clinical manifestations, laboratory findings, genetic test results and treatment information were extracted from patient files. For patients with multiple follow-up visits, the baseline visit at the time of diagnosis was used as the reference point for demographic and clinical variables. Clinical manifestations were defined based on documentation in the medical records during attack periods.
Clinical manifestations were categorised into musculoskeletal, gastrointestinal and systemic symptoms. Musculoskeletal manifestations included arthralgia, arthritis and myalgia. Gastrointestinal involvement was defined as recurrent abdominal pain suggestive of serositis during FMF attacks. Fever episodes and other systemic symptoms were also recorded when documented in the patient records. Symptoms were considered present if they were documented by the treating physician during clinical evaluation. Frequencies of these manifestations were analysed across different age groups.
Laboratory parameters were obtained from routine blood tests performed during attack periods or routine follow-up visits. These included WBC count, ESR, CRP and fibrinogen levels. Laboratory results were recorded as continuous variables and analysed in relation to clinical manifestations and genetic profiles.
Genetic analysis of the MEFV gene was performed using sequence analysis techniques. Peripheral blood samples were collected, and genomic Deoxyribonucleic acid (DNA) was extracted using standard procedures. Sequence analysis was conducted to identify common and rare MEFV mutations. Patients were classified according to mutation status as homozygous, heterozygous, compound heterozygous or mutation-negative. Particular attention was given to the presence of the M694V mutation due to its known association with disease severity.
Genetic testing for MEFV mutations was performed using peripheral blood samples obtained during routine clinical evaluation. DNA was extracted using standard extraction procedures according to the manufacturer’s instructions. The most common MEFV gene mutations were analysed using polymerase chain reaction-based methods and mutation-specific probes. Detected variants were classified according to previously described pathogenic mutations associated with FMF. Patients were categorised according to the presence of specific mutations, including M694V, E148Q, other mutations or the absence of detectable mutation. When more than one mutation was identified, the genotype was recorded accordingly.
Statistical Analysis
Statistical analyses were performed using standard statistical software. Continuous variables were expressed as mean ± standard deviation or median values, depending on data distribution, while categorical variables were presented as frequencies and percentages.
Comparisons between groups were performed using appropriate statistical tests according to the type of variable. Differences in laboratory parameters across age groups and sex were evaluated descriptively. A P <.05 was considered statistically significant.
Data obtained from the medical records of 786 patients treated for FMF between 2015 and 2020 were entered into Statistical Package for the Social Sciences software (version 22.0) for statistical analysis (IBM Corp., Armonk, NY, USA). The normality of data distribution was assessed using the Kolmogorov-Smirnov test. Depending on data characteristics, statistical comparisons were conducted using the Mann-Whitney U test, Kruskal-Wallis test or chi-square test. A P value of <.05 was considered statistically significant.
The study protocol was approved by the Cumhuriyet University Clinical Research Ethics Committee (Decision No: 2020–07/44, 8 July 2020) and was conducted in accordance with the Declaration of Helsinki. Due to the retrospective design and anonymised data collection, the requirement for written informed consent was waived by the Ethics Committee.
Results
Demographic Characteristics
Among the 786 patients with FMF, 527 (67.05%) were female, and 259 (32.95%) were male. The mean age of female patients was 38.28 ± 13.16 years (range: 18–73), while the mean age of male patients was 36.81 ± 11.86 years (range: 18–75) as only adult patients (≥18 years) were included in the study. The mean age at diagnosis was 28.30 years. The number of affected family members did not differ significantly between female (1.95 ± 1.79) and male patients (2.00 ± 1.81; P > .05).
Information on patient ethnicity was not systematically recorded in the clinical database. However, the study was conducted in a tertiary rheumatology centre in Türkiye, and the vast majority of patients were of Turkish origin.
Clinical Manifestations During Attacks
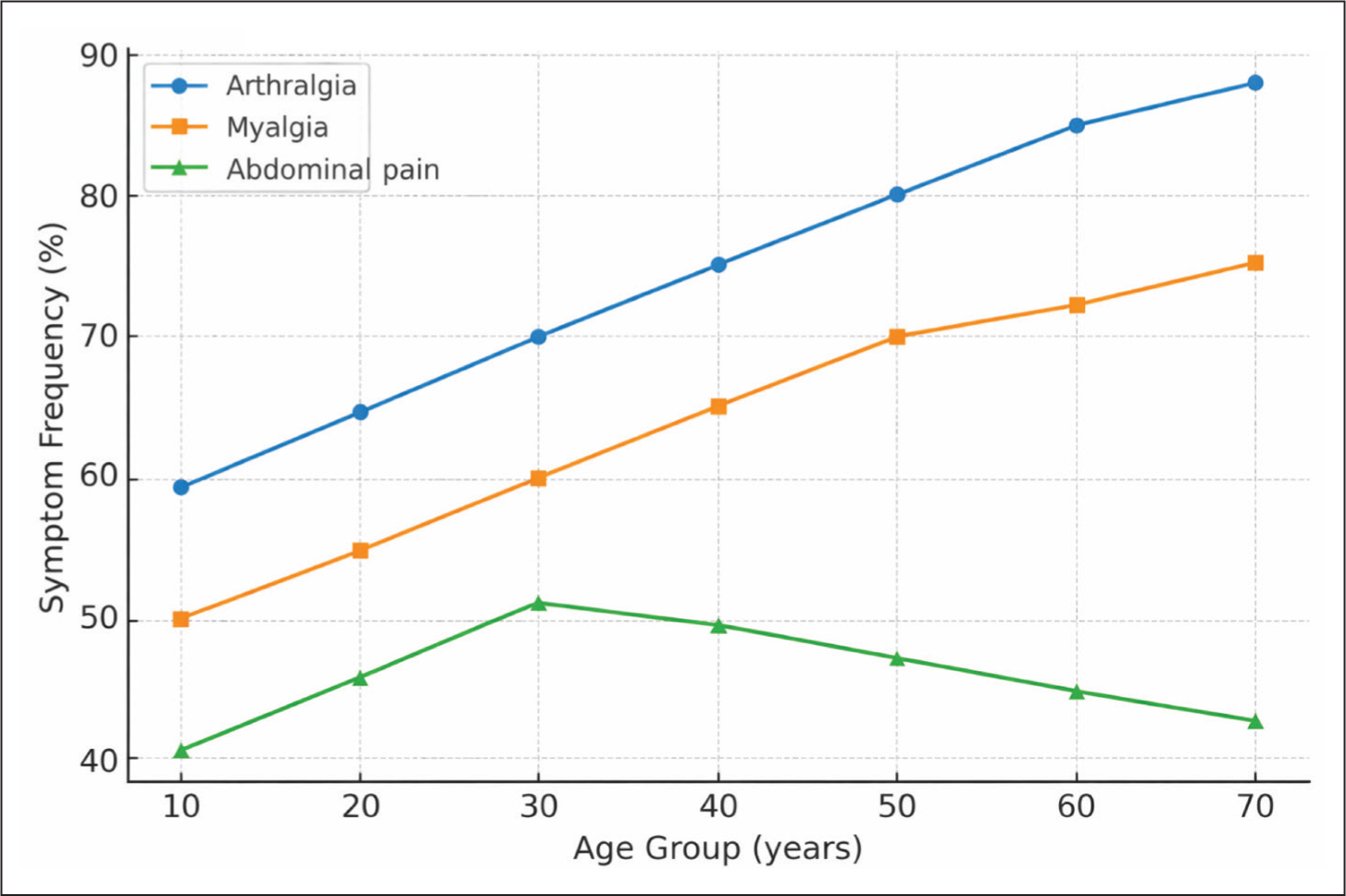
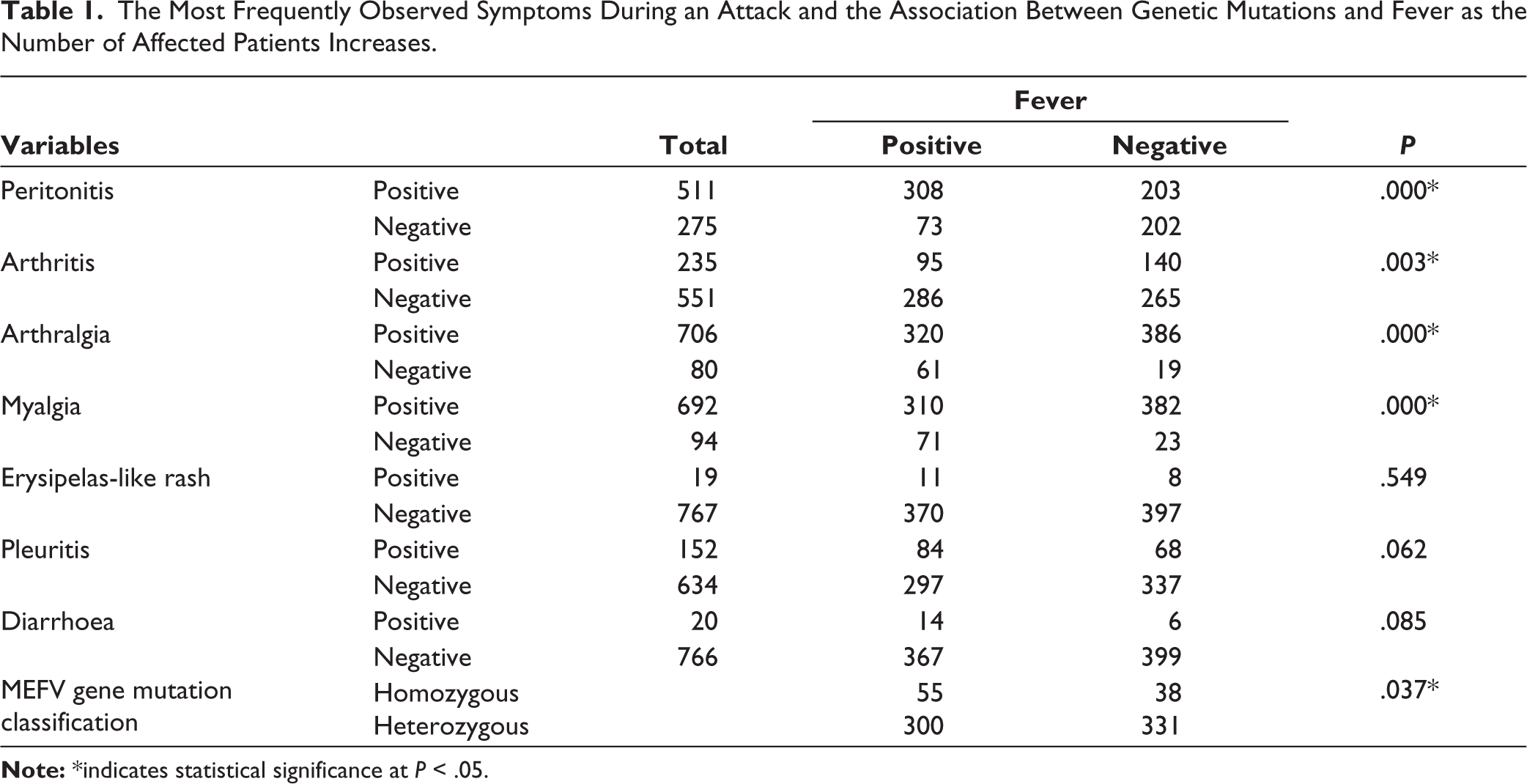
The most frequently observed symptoms during FMF attacks were arthralgia (89.82%), myalgia (88.04%), abdominal pain (65.01%) and fever (48.47%).
The distribution of clinical symptoms across age groups is illustrated in Figure 1.
Distribution of Symptoms by Age.
MEFV Genotype Distribution and Genotype-phenotype Associations
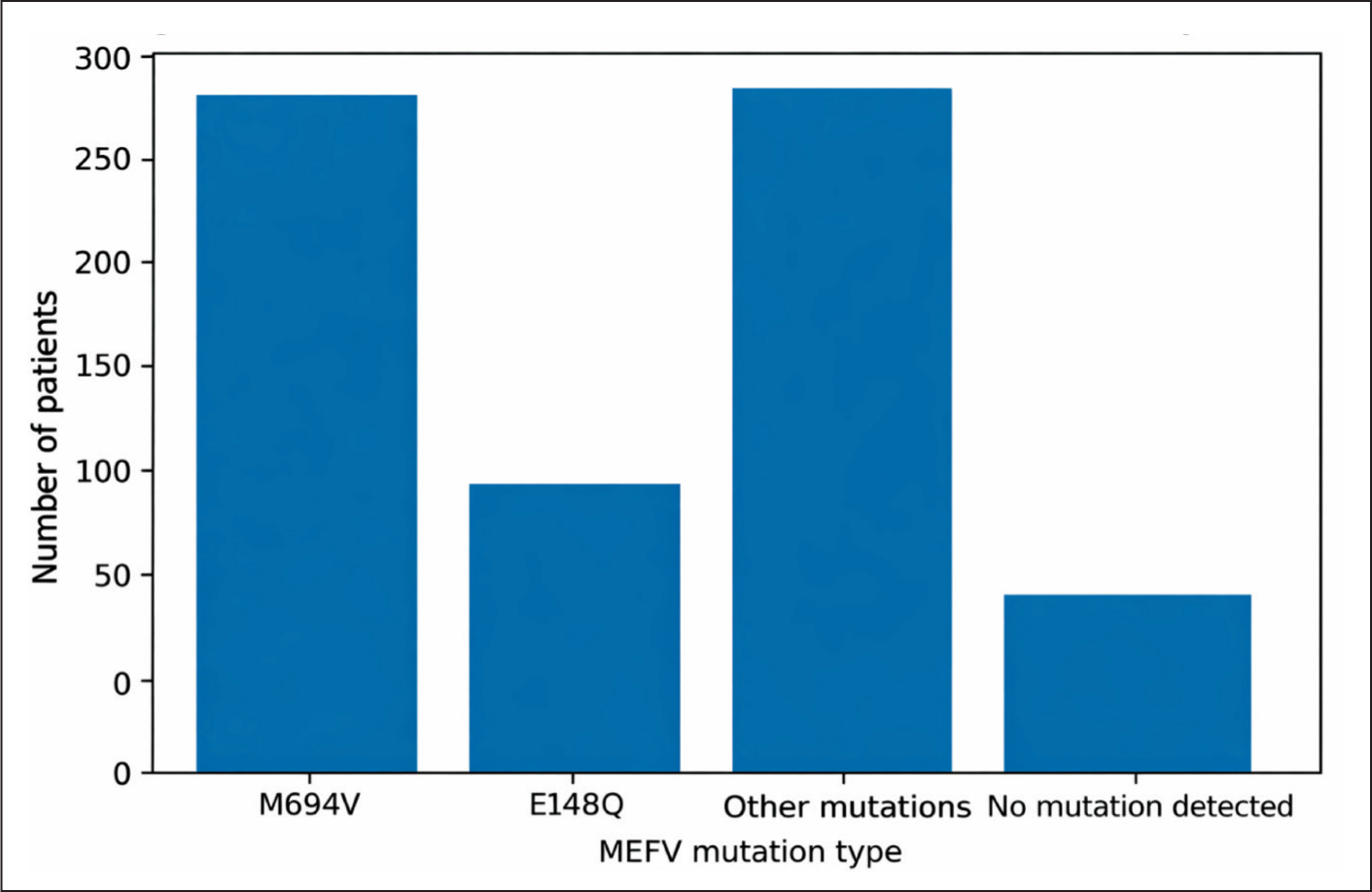
Homozygous MEFV mutations were identified in 93 patients (11.83%), while heterozygous mutations were detected in 631 patients (80.28%). Patients with homozygous mutations had a significantly higher number of affected family members compared to those with heterozygous mutations (P = .026).
Among genotype-phenotype associations, homozygous M694V mutations were significantly associated with higher frequencies of fever, peritonitis, arthralgia and myalgia compared to other mutation profiles (P = .037). In contrast, patients carrying V726A and E148Q mutations demonstrated milder clinical manifestations.
Analysis of the mutation spectrum showed that the most common mutation was M694V, detected in 309 patients (39.3%).
The distribution of the detected MEFV gene mutations in the cohort is illustrated in Figure 2.
Distribution of MEFV Gene Mutations in the Study Cohort.
In the clinical database used for this retrospective study, genetic results were recorded primarily as single detected mutations (heterozygous or homozygous) and compound heterozygous mutations were therefore not analysed as a separate category.
Clinical data were retrieved from electronic medical records and hospital databases by two independent investigators. When discrepancies were encountered, patient records were reviewed jointly, and consensus was reached. Disease manifestations were evaluated based on documented clinical findings during attacks. Musculoskeletal manifestations included arthralgia, arthritis and myalgia, while serosal involvement comprised abdominal pain, pleuritis and pericarditis.
For laboratory evaluation, acute-phase reactants measured during attack periods were preferentially included in the analysis. When multiple measurements were available, the value corresponding to the documented attack period was selected. These laboratory parameters were analysed in relation to genotype, sex, age groups and treatment response.
Treatment Response and Colchicine Resistance
At the time of evaluation, all patients were receiving colchicine therapy. A total of 718 patients (91.4%) responded to colchicine, whereas 68 patients (8.6%) were classified as colchicine-resistant.
Among colchicine-resistant patients, 37 (4.7%) were treated with canakinumab and 31 (3.9%) with anakinra. Colchicine resistance was significantly more frequent in patients with homozygous M694V mutations (P = .021) and this group demonstrated a higher requirement for interleukin-1 inhibitor therapy (P = .003).
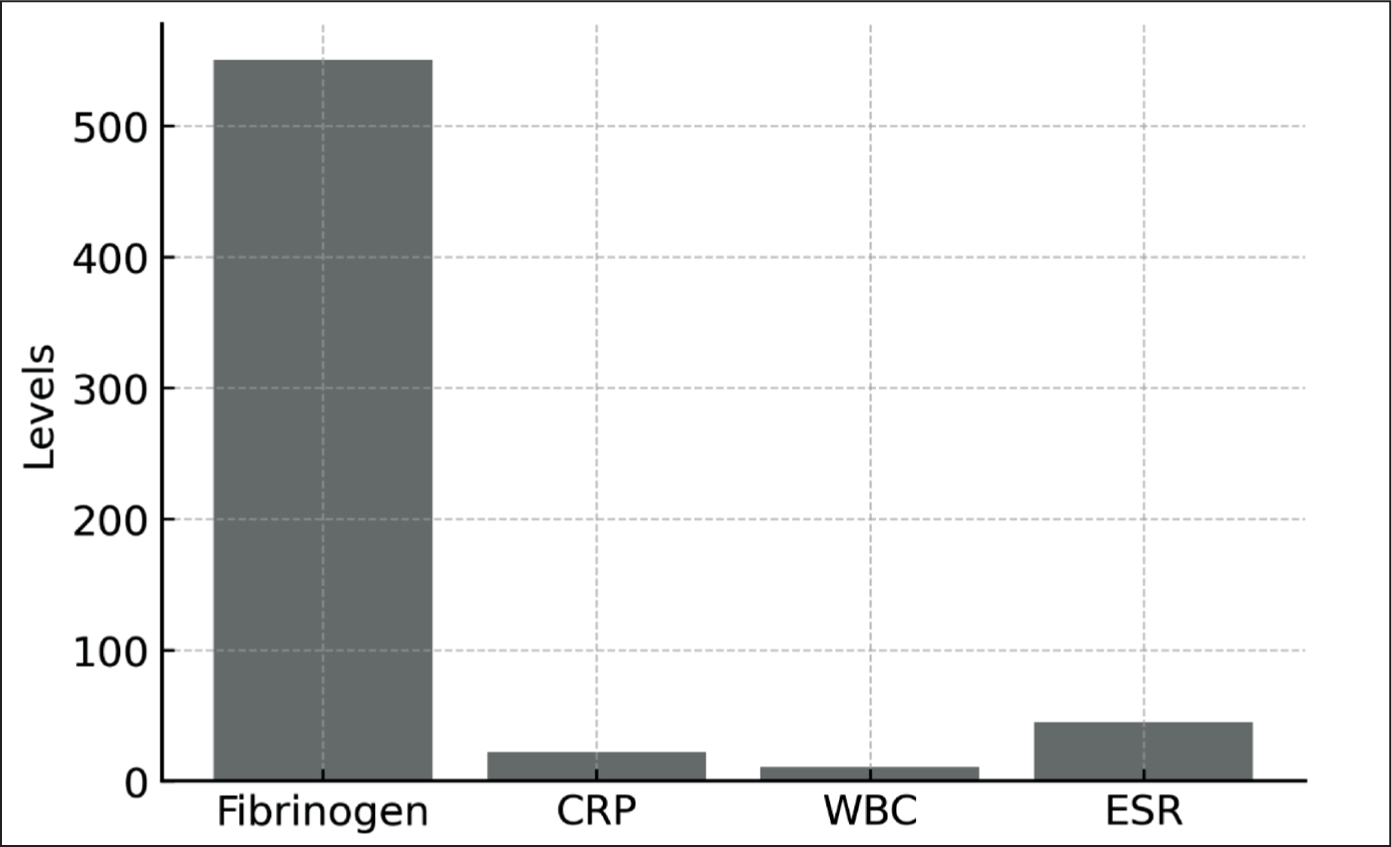
Laboratory parameters shown in Figure 3 represent measurements obtained during a single documented attack per patient at the time of clinical evaluation. Laboratory parameters were evaluated using measurements obtained during documented attack periods whenever available. Acute-phase reactants included ESR, CRP, WBC and fibrinogen levels. These laboratory parameters were analysed to assess the inflammatory burden associated with FMF attacks. When multiple measurements were available, the value corresponding to the attack period was selected for analysis. Laboratory values were also evaluated according to age groups and sex.
Inflammatory Markers During Attacks.
Acute-phase Reactants
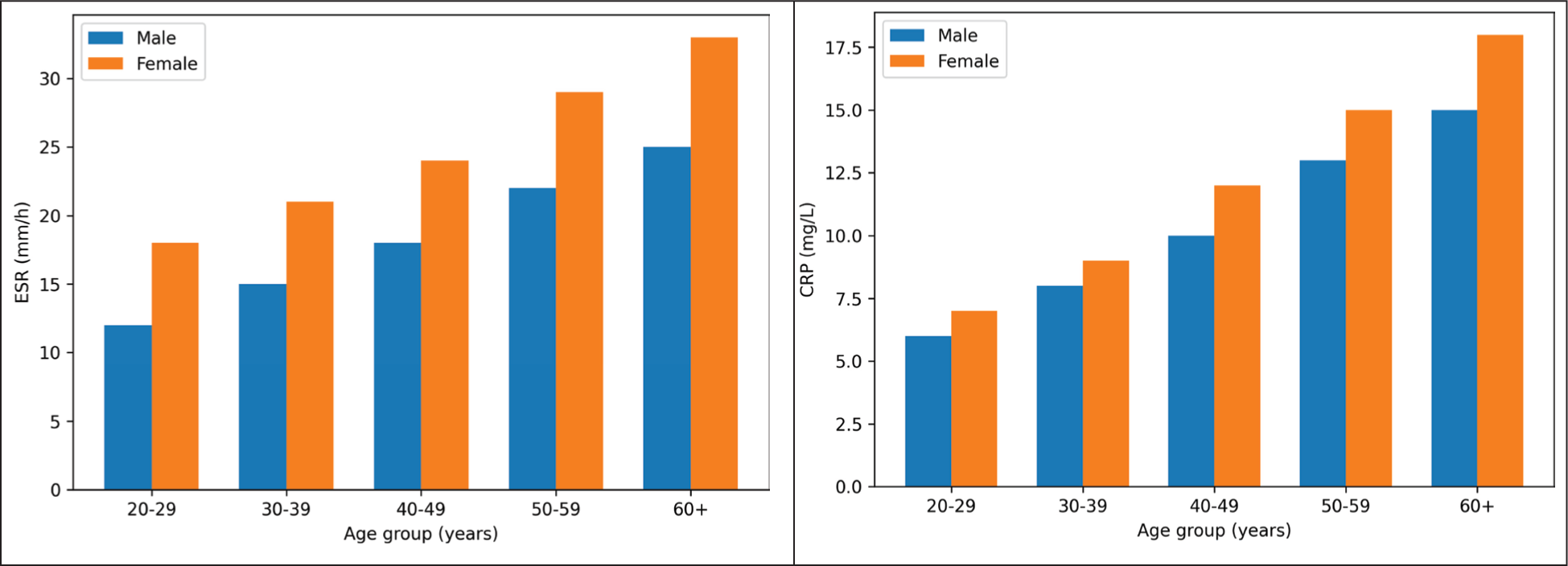
Figure 4a presents ESR levels stratified by age and sex, whereas Figure 4b illustrates CRP levels (P = .061). ESR values were significantly higher in female patients compared to males. CRP levels tended to be higher in patients presenting with arthritis during attacks, but this difference was not statistically significant (P = .089).
(a) ESR by Age and Sex. (b) CRP by Age and Sex.
Autoimmune Comorbidities and HLA Associations
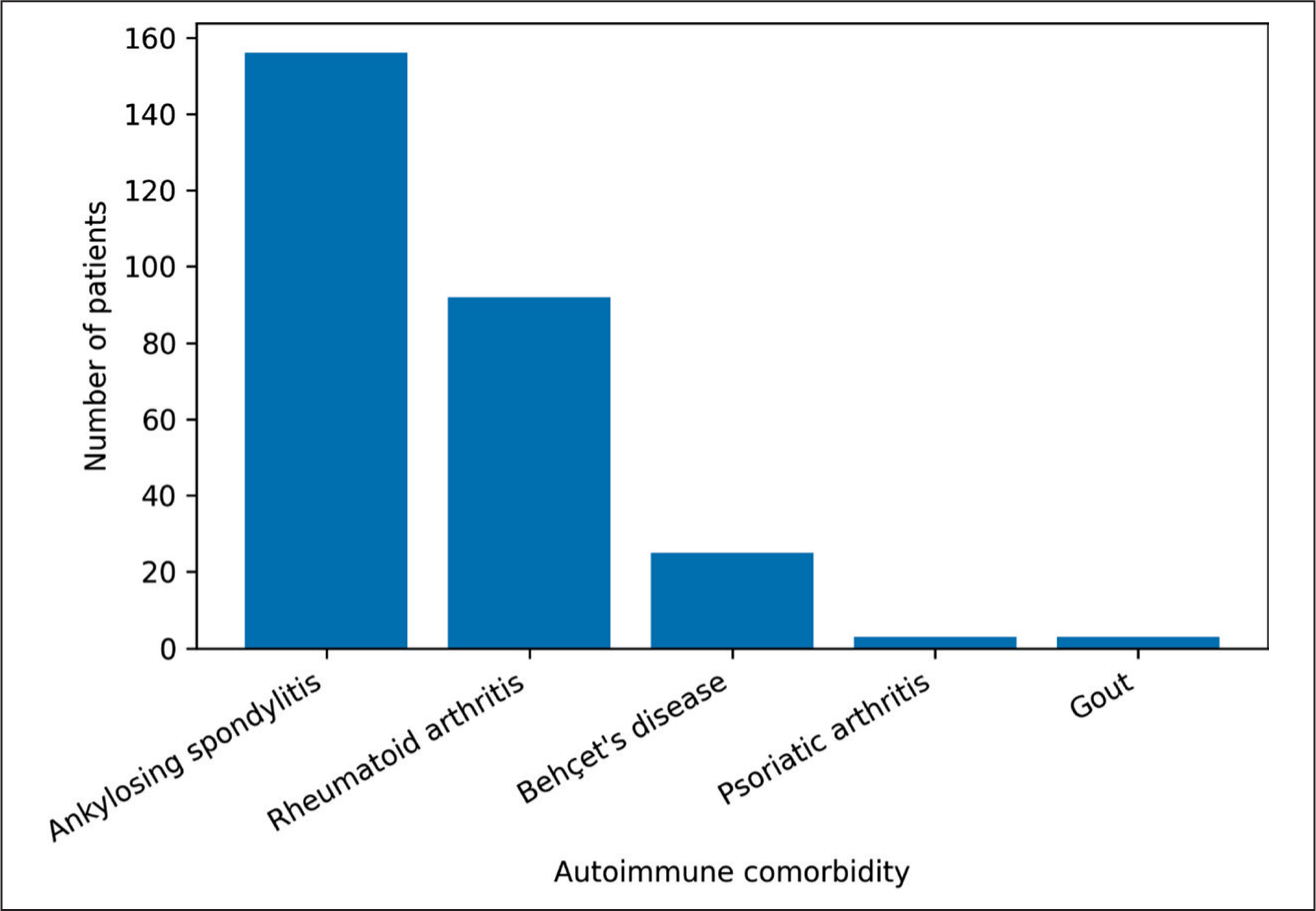
Comorbid autoimmune diseases were identified in 239 patients (30.4%). AS was the most frequent comorbidity, observed in 156 patients (19.84%), followed by RA in 92 patients (11.7%). Psoriatic arthritis and gout were each identified in three patients (0.38%). The distribution of autoimmune comorbidities in the study cohort is shown in Figure 5. Among patients with both FMF and AS, 71 (45.5%) were human leucocyte antigen (HLA)-B27 positive and 52 (33.3%) were HLA-B27 negative. HLA-B51 positivity was observed in nine (36%) of the 25 patients with concomitant FMF and Behçet’s disease. CRP levels were higher in HLA-B27-positive patients, although the difference was not statistically significant (P = .064).
Autoimmune Comorbidity in the FMF Cohort.
Discussion
The present study provides a comprehensive overview of genotype-phenotype relationships and autoimmune comorbidities in a large adult cohort of patients with FMF. Our findings highlight the heterogeneity of FMF manifestations and underscore the influence of both genetic background and coexisting autoimmune diseases on disease expression and treatment response.
Our findings confirm several previously established aspects of FMF while also providing new insights from a large adult cohort. In addition to confirming these well-known patterns, our study provides further information on the spectrum of autoimmune comorbidities observed in adult FMF patients and their relationship with genetic background and inflammatory markers.
FMF is classically characterised by recurrent inflammatory attacks driven by innate immune dysregulation, with MEFV mutations playing a central pathogenic role.16,17 Consistent with previous large cohort studies, musculoskeletal and serosal manifestations constituted the dominant clinical spectrum in our population. 18 Arthralgia and myalgia were particularly prominent, supporting the notion that musculoskeletal involvement represents a major source of disease burden in adult FMF patients. In line with prior reports, fever was not universally present during attacks, emphasising that its absence does not exclude active disease.16,19
Genotype-phenotype correlations remain a cornerstone of FMF research. Our findings reinforce the established association between M694V homozygosity and a more severe clinical phenotype, including higher inflammatory burden and suboptimal response to colchicine therapy.17–19 Similar observations have been reported in both Mediterranean and non-Mediterranean populations, suggesting that the pathogenic impact of this mutation transcends ethnic boundaries. 20 Conversely, mutations such as V726A and E148Q were associated with milder disease expression, consistent with earlier studies describing more favourable clinical courses in these genotypes.16,21
Treatment response patterns observed in our cohort further support the clinical relevance of genetic stratification. Colchicine resistance was predominantly observed in patients with high-risk genotypes, particularly M694V homozygosity, aligning with previous reports that identified this mutation as a predictor of treatment refractoriness. 22 The increased requirement for interleukin-1-targeted therapies in this subgroup mirrors real-world data from national and multicenter experiences, underscoring the need for individualised treatment strategies in severe FMF phenotypes. 23
The observed genotype distribution in our cohort also reflects the well-described mutation spectrum reported in Mediterranean populations. M694V remains the most prevalent mutation and has consistently been associated with more severe clinical manifestations. The relatively high frequency of this mutation in our cohort likely reflects the genetic background of the regional population. These findings support the concept that genotype distribution may vary across populations but that certain mutations maintain a consistent association with disease severity and treatment response.
A notable aspect of this study is the high prevalence of autoimmune comorbidities, particularly AS and RA. The coexistence of FMF with other inflammatory and autoimmune diseases has been increasingly recognised, suggesting shared inflammatory pathways and genetic susceptibility.24–27 Our findings are in agreement with prior reports documenting associations between FMF and Behçet’s disease, systemic lupus erythematosus and psoriasis.25–28 The observed HLA associations, although not reaching statistical significance in terms of inflammatory markers, further support the concept of overlapping immunogenetic mechanisms in these conditions.
The associations between fever, clinical manifestations and MEFV mutation status are summarised in Table 1. Several studies have suggested that MEFV mutations may contribute to an enhanced inflammatory response through dysregulation of the pyrin inflammasome, potentially facilitating the development of other immune-mediated inflammatory diseases. In particular, the coexistence of FMF and AS has been described in multiple cohorts from Mediterranean countries. Some investigators have proposed that MEFV mutations may act as disease-modifying factors in spondyloarthritis by amplifying IL-1-mediated inflammatory pathways. Although the exact mechanisms remain incompletely understood, these observations support the hypothesis that autoinflammatory and autoimmune diseases may share partially overlapping pathogenic pathways.
The Most Frequently Observed Symptoms During an Attack and the Association Between Genetic Mutations and Fever as the Number of Affected Patients Increases.
Our findings are consistent with these observations, as AS represented the most frequent autoimmune comorbidity in our cohort. This high prevalence may reflect both shared genetic susceptibility and increased clinical recognition of spondyloarthritis manifestations in FMF patients. From a clinical perspective, the coexistence of these conditions may complicate diagnostic evaluation and influence treatment decisions, particularly in patients with persistent musculoskeletal symptoms despite adequate colchicine therapy.
Another noteworthy observation from our cohort is the broad spectrum of clinical manifestations observed in adult FMF patients. Although FMF is traditionally described as a disease characterised by recurrent febrile serositis, our findings indicate that musculoskeletal symptoms such as arthralgia and myalgia represent major components of the clinical burden in adult populations. These findings are consistent with previous reports suggesting that articular and periarticular manifestations are common in FMF and may significantly affect quality of life. Recognition of these manifestations is particularly important in adult rheumatology practice, where FMF may present with atypical or incomplete attack patterns.
Another important aspect highlighted by our findings is the clinical relevance of screening for autoimmune comorbidities in FMF patients. In routine clinical practice, symptoms such as persistent arthritis, axial pain or systemic inflammatory manifestations may sometimes be attributed solely to FMF activity. However, the coexistence of distinct autoimmune diseases may require different therapeutic approaches. Early recognition of overlapping conditions may therefore improve patient management and optimise treatment strategies. For example, patients with concomitant AS may benefit from biologic therapies targeting tumour necrosis factor or IL-17 pathways, whereas patients with refractory FMF attacks are more likely to require IL-1-targeted treatments. These observations underscore the importance of a comprehensive rheumatologic evaluation in patients with FMF, particularly in adult populations where disease presentation may be more heterogeneous than in paediatric cohorts.
The strengths of this study include its large sample size and detailed characterisation of clinical, genetic and immunological features in an adult FMF population. However, several limitations should be acknowledged. The retrospective design and reliance on baseline data precluded longitudinal assessment of disease outcomes, survival and long-term complications such as amyloidosis. In addition, the single-centre nature of the study may limit the generalisability of the findings. Nevertheless, the comprehensive baseline analysis provides valuable insights into disease heterogeneity and real-world treatment patterns.
In conclusion, this study highlights the complex interaction between genetic background and coexisting autoimmune conditions in adult FMF patients. Our findings emphasise that FMF is not only a monogenic autoinflammatory disorder but also a condition that may coexist with a broad spectrum of immune-mediated diseases. Recognition of these overlaps is important for clinical management and may help guide individualised therapeutic strategies. Future prospective and multicenter studies are needed to further clarify the mechanisms underlying these associations and to evaluate their impact on long-term outcomes.
Footnotes
Acknowledgements
The authors would like to thank all participants and clinical staff who contributed to this study. The authors confirm that no medical writing or editorial assistance from a third party was used in the preparation of this manuscript.
Authors’ Contribution
The concept and initial study design were developed by Ali Şahin. Betül Sarı Kalın collected the core data as part of her thesis. Neşe Çabuk Çelik transformed the thesis into a publishable manuscript and led the preparation of the final article. Rümeysa Yılmaz Göç contributed to figure design and critically reviewed the manuscript during the writing process. Serpil Albayrak supported data collection and organisation. All authors contributed to the interpretation of findings and approved the final version of the manuscript.
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author upon reasonable request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical Approval
This retrospective cohort study was conducted at the Division of Rheumatology of a university hospital and received approval from the Institutional Ethics Committee (Decision No: 2020–07/44, dated 8 July 2020). Patients who presented to the outpatient clinic between 2015 and 2020 and were diagnosed with FMF according to the Tel-Hashomer criteria were included. All participants or their legal guardians provided written informed consent before genetic testing, in accordance with the Declaration of Helsinki.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Informed Consent
Informed consent was waived by the Ethics Committee due to the retrospective design of the study and the use of anonymised data.