
Abstract

Introduction
Blau syndrome (BS) was first described in 1985 by Edward Blau and is a rare autosomal dominant disease characterised by the triad of arthritis, uveitis and dermatitis in early childhood. 1 The BS locus was later mapped to chromosome 16q12.1-13 by Tromp et al., and Miceli-Richard et al. subsequently identified the mutations in the CARD15/NOD2 gene. 1 BS is an exceptionally rare autoinflammatory disorder, with an estimated prevalence of 0.05 per 100,000 person-years. 2 We herein report a case of BS.
Case Presentation
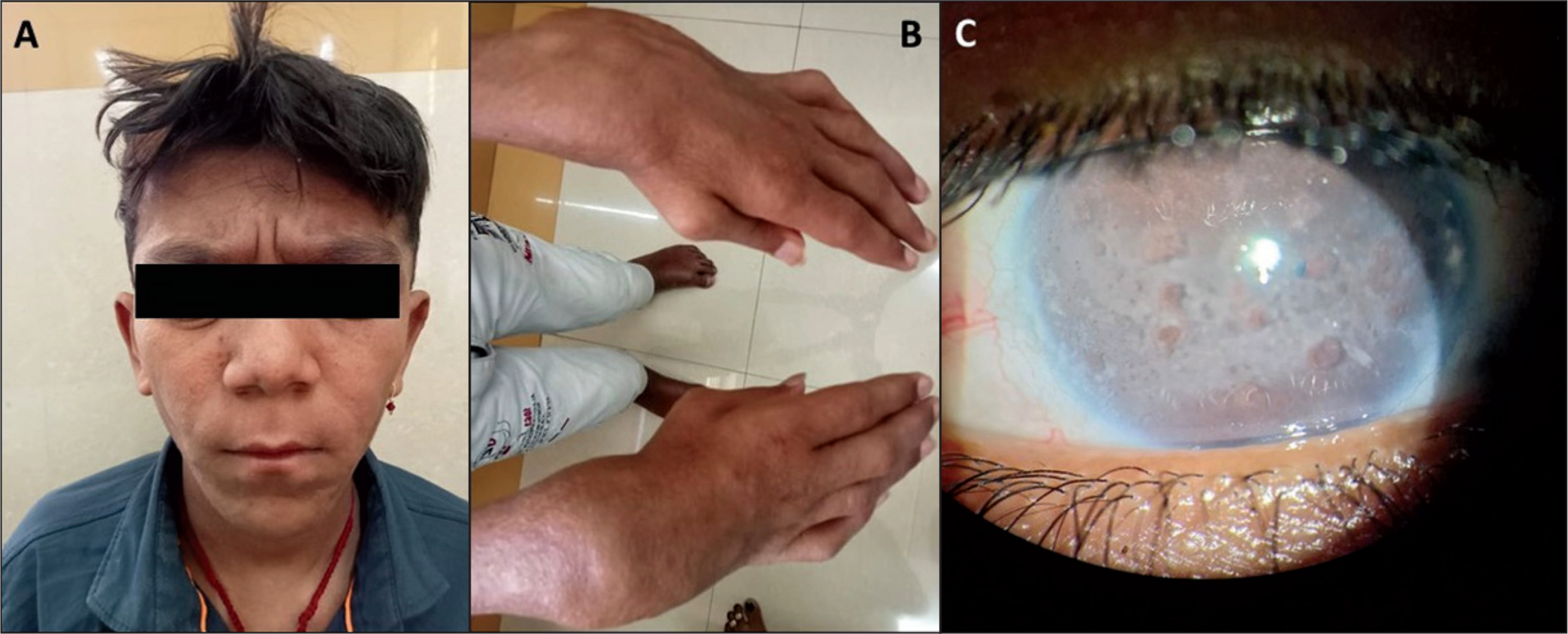
A 9-year-old boy, born to a non-consanguineous couple with no family history of autoimmune disease, presented with a skin rash since 6 months of age. At 2 years of age, he developed bilateral wrist joint pain and swelling. Over the past 6 months, he has also developed reddish discolouration of the eyes with blurring of vision. Examination revealed a coarse face (Figure 1A) with a photosensitive maculopapular rash over the whole body with nasolabial sparing. He also had boggy swelling over the wrist, camptodactyly and polyarthritis (wrist, knee and small joints of the hands) (Figure 1B). Boutonniere deformity was noted in the bilateral thumbs. Ophthalmologic examination showed bilateral band-shaped keratopathy (Figure 1C), anterior uveitis and glaucoma with an Intra Ocular Pressure of 26 mmHg in both eyes (Normal: 12-21 mmHg). Systemic examination revealed a liver 2 cm below the right costal margin. Hypogonadism was also seen. On evaluation, hemogram, ESR, CRP, lipid profile, thyroid, renal and liver function tests were normal. Rheumatoid factor, ANA(IF) and ANA profile were also negative. Urine routine and urine glycosaminoglycans were normal. Ultrasound of the abdomen and pelvis revealed hepatosplenomegaly with bilateral small testes.
Coarse Facies with Maculopapular Rash (A) and Boggy Swelling of Wrist, Camptodactyly with Deformities (B), Slit Lamp Images of Both Eyes Showing Conjunctival Congestion, Band-shaped Keratopathy, KPs, Shallow AC (C).
Genetic analysis using next-generation sequencing (NGS) identified a heterozygous missense variant in the NOD2 gene: c.919C>T (p.Arg307Trp), corresponding to chr16:g. 50744822C>T (GRCh37). This substitution of arginine with tryptophan at codon 307 is absent from gnomAD, suggesting it is not a common polymorphism. In silico prediction tools indicated a deleterious effect (REVEL 0.731, CADD 25.8), and ClinVar has classified the variant as pathogenic, supporting its relevance to BS. Although family members were not tested, the presence of the p.Arg307Trp variant in our patient is consistent with a likely de novo NOD2 mutation. The patient was finally diagnosed as a sporadic case of BS.
The child was started on Prednisolone 20 mg, which was gradually tapered and on low dose with ongoing azathioprine 2 mg/kg. All symptoms completely resolved with residual deformity, and he continues to be on follow-up.
Discussion
BS is underreported in India, with approximately 30 genetically confirmed cases. Janarthanan et al. reported the first molecularly confirmed familial BS in India. 3 The largest South Indian case series includes seven patients, reported by Babu and Rao. 4 Kumarah et al. outlined 11 cases from 10 families, which was the largest North Indian cohort. 5 BS consists of the classical triad of dermatitis, arthritis and uveitis. The rash is usually the first to appear (first year of life), followed by arthritis (2-4 years of age) and lastly uveitis (around 4 years of age). 5 The median age of ocular disease is 4.4 years (range = 6 months-22 years). 6 In accordance, our patient developed a maculopapular rash at 6 months of age, joint involvement was seen at 2 years of age, with ocular involvement at around 9 years of age. Fever is an important manifestation of BS in childhood, although it is not included in the classical triad. 7
Two main patterns of skin involvement are seen in BS: rash and multiple subcutaneous nodules. 8 Rash is described as a maculopapular, brownish rash which may be tender. Pinhead-sized, lichenoid, yellow to brown papules appear in clusters, which may become confluent. Also, multiple firm subcutaneous plaques or nodules may be present, which are only apparent on palpation. The appearance is variable, with some cases having erythematous eruptions while others have intermittent and/or generalised eruptions, which are often seen on the trunk and/or extremities.1,8 Biopsy of the skin lesions shows non-caseating granulomas with multinucleated giant cells. 1
Joint involvement in BS is mostly in the form of symmetric polyarthritis involving the wrists, metacarpophalangeal (MCP), first metatarsophalangeal (MTP) and proximal interphalangeal (PIP) joints of the hands and feet, ankles and sometimes elbows and knees. 1 Joint effusion due to granulomatous synovitis and tenosynovitis is seen. 2 Arthritis may be chronic and can lead to deformities of the fingers like camptodactyly (fixed flexion deformity of a finger at the PIP joint) and Boutonniere deformity (PIP is flexed while the distal interphalangeal joint is hyperextended) as seen in our case.1,8 Camptodactyly was also described by Blau in his cohort of a family of 11 members from four generations who were affected with granulomatous disease of the skin, eyes and joints. 9 Although arthritis has a chronic course, destruction or erosion of the joints is usually not seen in BS.1,8
Ocular manifestations of BS are seen in over 60%-80% of patients and are usually bilateral. 6 The most common manifestation is granulomatous uveitis.1,5,6,8 Uveitis most frequently presents as posterior uveitis, which can potentially become panuveitis with multifocal choroiditis. 10 Unlike JIA, where anterior uveitis is seen, there are reports which suggest panuveitis is seen in around 75% of BS patients. 10 Other manifestations include papilledema, keratitis, episcleritis, conjunctivitis and corneal ulcers. Over time, untreated cases may lead to long-term complications like cataract, glaucoma, band keratopathy, posterior synechiae, macular oedema, retinal detachment, choroidal thickness reduction, chorioretinal scars, choroidal neovascularisation and optic nerve atrophy. 2
Extra-triad manifestations of BS include fever, erythema nodosum, sialadenitis, leucocytoclastic vasculitis, neuropathies, granulomatous glomerular and interstitial nephritis, interstitial lung disease, pericarditis, pulmonary embolism, hepatic granulomas, splenic involvement, pulmonary and systemic hypertension, lymphadenopathy and hepatosplenomegaly. 10
BS is caused by a heterozygous pathogenic variant in the CARD15/NOD2 gene which is located on chromosome 16q12. 2 NOD2 genetic mutations lead to activation of the NF-κβ pathway, leading to an overproduction of inflammatory cytokines like IL-1β, TNFα and IL-6 which drive the formation and maintenance of granulomas,2,7 Most of the mutations in BS are seen in the NACHT domain which results in the NF-κβ activation as discussed above. 11 The most common mutation is the substitution of arginine at position 334 by glutamine (R334Q) or tryptophan (R334W).5,8 To our knowledge, the R307W genetic variant (substitution of arginine with tryptophan at codon 307 of the NOD2 protein) found in our case is one of the first documented cases in the literature. This case contributes to the expanding mutational spectrum of NOD2 and reinforces the pathogenicity of non-hotspot variants in BS.
Table 1 shows a summary of hotspot and non-hotspot NOD2 variants and their associated phenotypes.
Genotype-phenotype Correlation: Summary of Hotspot and Non-hotspot NOD2 Variants and Their Associated Phenotypes.

The limitation of this report is to prove the causality of the novel variant
Conclusion
This case highlights the importance of considering BS in children with arthritis, uveitis and skin findings.
It underscores the clinical utility of genetic/molecular testing in diagnosing BS and differentiating it from other inflammatory and syndromic mimics.
It also adds to the growing body of evidence for rare NOD2 mutations like R307W.
Footnotes
Authors’ Contribution
Yashas BP, Shiva Prasad BN and Deepa Bhat contributed to conceptualisation. Shiva Prasad BN, Ramaswamy Subramanian and Mahabaleshwar Mamadapur contributed to supervision. Yashas BP, Shiva Prasad BN and Deepa Bhat contributed to writing - original draft. Yashas BP, Deepa Bhat, Shiva Prasad BN, Ramaswamy Subramanian and Mahabaleshwar Mamadapur contributed to writing - review and editing. All authors approved the final version of the manuscript and take full responsibility for its accuracy and integrity.
Declaration of Conflicting Interests
Dr Mahabaleshwar M is associate editor and Dr Subramanian R is national editorial member of this Journal. They have not been involved in the peer review of this article.
Ethical Approval
Not applicable.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Patient Consent
Written informed consent to publish was obtained from the patient.