
Abstract

Dear Editor,
A 55-year-old man presented with progressively worsening respiratory symptoms. He reported exertional dyspnoea for 2 years, with marked worsening over the preceding 4 months, accompanied by a persistent dry cough. He denied fever, weight loss, arthralgia, myalgia or cutaneous rashes. There was no history of smoking, occupational or environmental exposure or contact with birds or mould.
The patient reported a 2-year history of recurrent dry, scaly fingertip lesions with occasional fissuring and mild discomfort, involving the lateral and palmar aspects of the fingers, predominantly the radial border of the middle finger and the tip of the ring finger. He had partial relief with emollients and topical corticosteroids, and there were no features suggestive of systemic autoimmune disease, including Raynaud’s phenomenon, dysphagia or sicca symptoms.
On examination, vital parameters were normal. Cutaneous examination revealed dry, fissured skin over the fingers, raising suspicion of subtle mechanic’s hands (Figure 1A). Pulmonary examination revealed fine bibasilar inspiratory crackles, more prominent on the right. Other systemic examinations were unremarkable.
(A) Mechanic’s Hands with Hyperkeratosis Over the Radial Aspect of the Middle Finger and Fissuring at the Tip of the Ring Finger. (B-C) HRCT Chest Showing Axial (B) and Coronal (C) Images with Fibrotic NSIP Pattern, Characterised by Bilateral Basal Reticulation with Traction Bronchiectasis (Right > Left) and Ground-glass Opacities in the Left Lower Lobe, with Minimal Honeycombing.
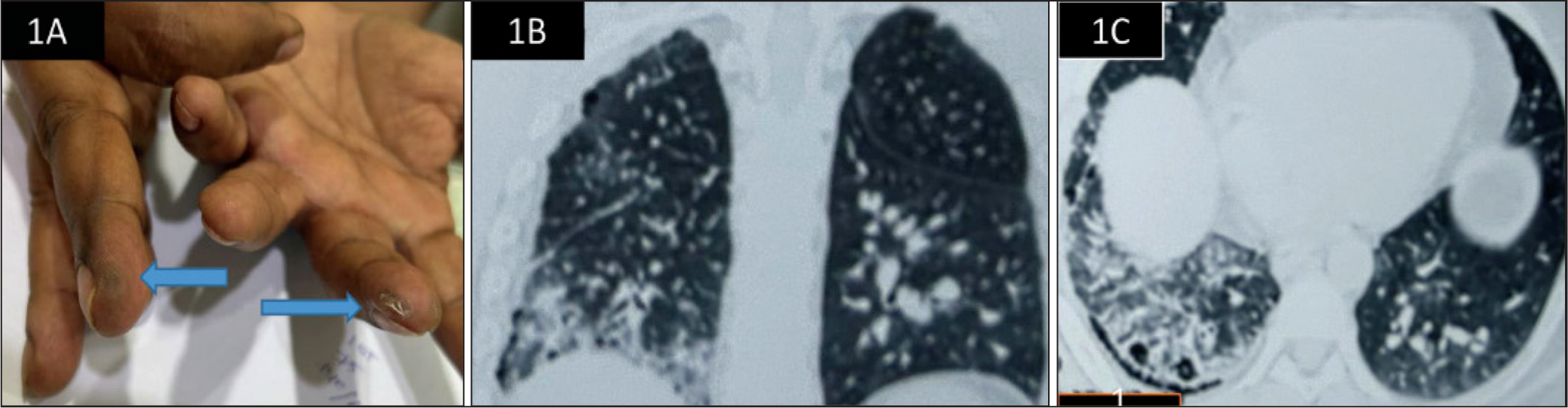
Laboratory investigations, including complete blood counts, renal and liver function tests, thyroid profile and muscle enzymes, were within normal limits. Chest radiograph showed bilateral basal opacities. Nailfold capillaroscopy was normal. High-resolution computed tomography of the thorax revealed a usual interstitial pneumonia (UIP) pattern with subpleural reticulation, minimal honeycombing and traction bronchiectasis, predominantly in the right lower lobe, with ground-glass opacities in the left lower lobe (Figure 1B-1C).
Pulmonary function testing revealed a moderate restrictive ventilatory defect with total lung capacity at 62% of predicted and a reduced diffusing capacity (DLCO) at 47% of predicted.
Autoimmune evaluation revealed anti-Jo-1 antibody positivity on an ENA immunoblot panel, while anti-cyclic citrullinated peptide antibodies, assessed by ELISA, were negative; rheumatoid factor was also negative. In the absence of clinical or biochemical evidence of myositis or arthritis, a diagnosis of anti-Jo-1 antisynthetase syndrome (ASSD) presenting as isolated interstitial lung disease was made. The patient was treated with oral prednisolone (0.5 mg/kg/day) and mycophenolate mofetil (1 g twice daily), resulting in significant symptomatic improvement, stabilisation of pulmonary function at 3 months and complete resolution of mechanic’s hands in parallel with respiratory improvement.
Discussion
ASSD is an autoimmune disorder defined by anti-aminoacyl-tRNA synthetase antibodies, most commonly anti-Jo-1, classically associated with ILD, myopathy and arthritis. 1 In clinical practice, ILD may be the earliest manifestation and can remain the sole presenting feature for a prolonged period, with muscle or joint involvement developing later or remaining absent.1,2
Non-specific interstitial pneumonia (NSIP) is the most common ILD pattern in ASSD, while UIP is less frequent.1,3 NSIP shows ground-glass opacities, homogeneous distribution, minimal honeycombing and subpleural sparing, whereas UIP demonstrates basal predominance, reticulation, honeycombing and architectural distortion with an apico-basal gradient.³ Fibrotic NSIP is characterised by interstitial fibrosis with reticulation and traction bronchiectasis, often accompanied by ground-glass opacities and minimal or absent honeycombing³ ILD predominant disease is more commonly associated with non-Jo-1 antibodies, while the absence of myositis in anti-Jo-1 positive patients is uncommon.4,5 This case highlights that anti-Jo-1 positive ASSD can present as a fibrotic NSIP/UIP pattern ILD without clinical myositis and should be considered in the differential diagnosis of idiopathic ILD. Recognition of this presentation is important, as early initiation of immunosuppressive therapy may help preserve lung function and improve outcomes.1,5
Footnotes
Consent for Publication
Written informed consent for publication of the clinical details and images was obtained from the patient.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical Approval
NA.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.