
Abstract
Supersalts are ionic compounds formed by pairing superalkali cations with superhalogen anions, resulting in systems with strong charge-transfer characteristics. This study investigates novel supersalts of the form H2M3–Y (M = Li or Na; Y = AlF4, BeF3, NO3) using the MP2/aug-cc-pVTZ level of theory. Their stability was evaluated via binding energy (Eb) and dissociation energy (Ed), with H2Li3–AlF4 showing the highest binding energy (11.35 eV), followed by H2Li3-BeF3 (10.66 eV) and H2Li3-NO3 (7.58 eV). Net charge analysis confirms significant charge separation (+0.93e / −0.92e in Li systems), supporting the ionic nature of these complexes, further validated by QTAIM analysis. The HOMO–LUMO gap is highest for H2Li3–BeF3 (10.33 eV), indicating relatively higher electronic stability. The results indicate that Li-based supersalts are more stable than their Na-based counterparts, with stability following the trend AlF4 > BeF3 > NO3. These findings provide a basis for understanding charge-transfer-driven molecular systems.
Introduction
Supersalts represent a distinct category of ionic compounds formed through the interaction of molecular species known as superatoms, which exhibit periodic element-like behavior. They are charge-transfer salts, comprising superalkali cations and superhalogen anions, similar to conventional ionic salts that contain alkali cations and halogen anions. However, they are distinct in their tendency to preferentially dissociate into ionic fragments, distinguishing them from traditional salts. Superalkalis, 1 characterized by ionization potentials lower than those of alkali metals, and superhalogens, 2 defined by their exceptionally high electron affinities, are prime candidates for forming such compounds. Khanna and Jena's 3 pioneering work revealed that specific atomic clusters, known as superatoms, can replicate elemental properties. Recent studies showcase their potential for creating novel materials. 4 This leads to a fascinating question: Can supersalts be crafted from the union of positively and negatively charged superatomic ions? Just as alkali metals and halogens combine to form conventional salts, the interaction of these two superatomic species leads to the development of supersalts. Several notable investigations have been conducted on supersalts. For instance, Li et al. 5 studied the interactions of the BLi6 superalkali with various superhalogens (LiF2, BeF3, and BeF4), while Yang et al., 6 explored how the BF4 superhalogen interacts with FLi2, OLi3, and NLi4 superalkalis. More recently, a theoretical study on supersalts Na2MgX4 (X = F, Cl) was conducted by exploring their formation via association reactions involving superalkalis, superhalogens, dimers, and conventional salts in both ionic and neutral forms. 7 Additionally, research has demonstrated that interactions between Li2X-based superalkalis and LiX2-based superhalogens lead to the formation of Li3X3 supersalts for X = F, Cl, Br, and I.8,9 Similarly, potassium-iodide clusters K2I and KI2 generate a superalkali cation (K2I⁺) and a superhalogen anion (KI2⁻), which combine to form the stable supersalt K3I3. 10 The series of supersalts, including (M3O)⁺(Al13)⁻ (M = Na, K), 11 and (Na2X)⁺(Y)⁻ (X = SCN, OCN, CN; Y = MgCl3, Cl, NO2), 12 have been theoretically designed and identified as promising inorganic NLO molecules due to their significant nonlinear optical responses. The superacids and superbases can also be used as building blocks of the supersalts. 13 Reportedly, classic superalkalis (FLi2, OLi3, NLi4), when interacting with superhalogens (BeF3, BF4, and NO3), can form salts while retaining the cationic and anionic nature of their components. 14 Recent theoretical studies highlight that excess-electron-driven interactions play a key role in stabilizing unconventional ionic and supramolecular systems. In particular, superalkali-assisted metalide complexes and vdW-stabilized assemblies demonstrate how long-range electrostatics, charge transfer, and noncovalent interactions govern electronic structure and stability, providing a broader framework for understanding charge-separated molecular architectures.15,16
In this work, we investigate supersalts derived from H2M3 (M = Li, Na) 17 superalkalis through direct pairing with superhalogens (AlF4, BeF3, and NO3). Unlike widely studied supersalt systems based on FLi2, OLi3, and NLi4, 14 this study focuses on supersalts formed from H2M3 units, which represent a comparatively less explored class. A systematic analysis of their stability, charge-transfer characteristics, and bonding nature is carried out, along with a comparison of Li- and Na-based systems to understand how the metal center influences interaction strength and overall stability. This also enables us to examine whether established supersalt formation trends remain valid for this class of systems.
Computational details
The equilibrium geometries were determined using second-order Møller-Plesset (MP2) perturbation theory,
18
with the basis set aug-cc-pVTZ in the Gaussian 16 software package.
19
For visualization purposes, we have used the GaussView package.
20
The MP2/aug-cc-pVTZ level was chosen for its reliable description of charge transfer and noncovalent interactions. Previous studies on superalkali-based and related charge-separated systems have successfully employed MP2-level calculations to describe interaction energies and structural properties.
17
Therefore, the present level is considered appropriate for a comparative analysis of these systems. While higher-level methods such as CCSD(T) or multi-level approaches may provide more accurate absolute energies, MP2 offers a reasonable balance between accuracy and computational cost. Accordingly, the present results are interpreted within a comparative framework, and the observed energetic trends are expected to be qualitatively reliable. Frequency calculations were performed at the same level of theory to confirm that all optimized structures correspond to true minima on the potential energy surface, as no imaginary frequencies were observed. The calculated vibrational spectra for the optimized structures are provided in the Supporting Information (Figure S1 to Figure S11). Atomic charges were obtained using Natural Population Analysis (NPA) within the Natural Bond Orbital (NBO) framework, which provides a more reliable description of charge distribution in molecular systems.
21
To gain deeper insight into the interactions, we performed Quantum Theory of Atoms in Molecules (QTAIM) analysis
22
using the Multiwfn package.
23
The energy gap (Egap), calculated as the difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), is a key parameter in understanding electronic properties and chemical reactivity. Their stability has been assessed based on binding and dissociation energy, which is determined using the following equations:
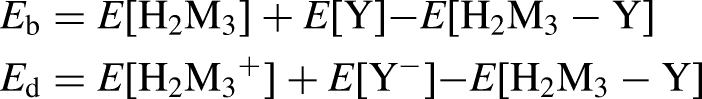
Results and discussion
In this section, we explore H2M3-Y (where M = Li and Na and Y = AlF4, BeF3, and NO3) supersalts. Our analysis begins with the H2M3-AlF4 (M = Li and Na) complexes. Figure 1 presents the optimized geometries of the ground-state and low-energy isomers of neutral H2M3-AlF4 (M = Li and Na), along with their corresponding bond lengths and NPA charges. The relative energy (Er) is listed in Table 1. The Er of the H2Li3-AlF4 (b) and H2Li3-AlF4 (c) are 0.04 eV and 0.06 eV, respectively, whereas the Er of H2Na3-AlF4 (b) is 0.33 eV, indicating relative stability compared to the lowest energy isomer (a) in each case.
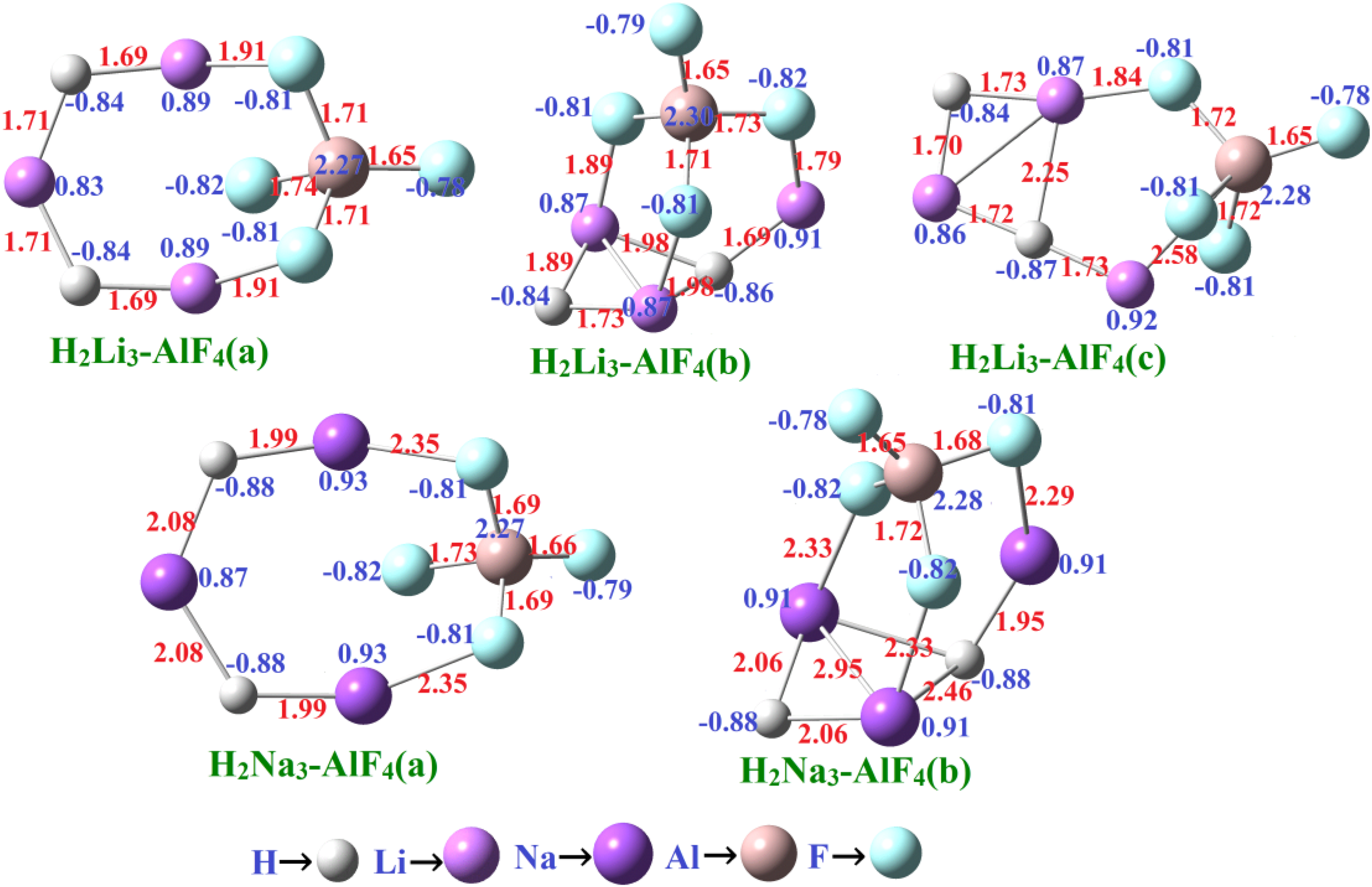
Optimized geometries of isomers H2M3-AlF4 (M = Li and Na) with selected bond lengths in Å and net charge at the MP2/aug-cc-pVTZ method.
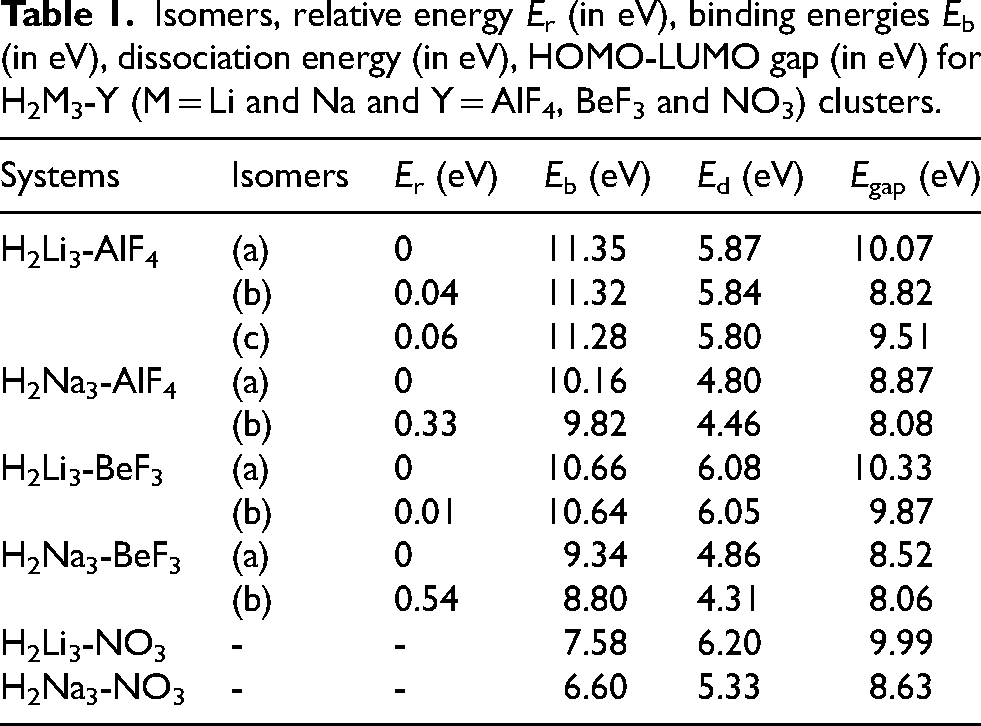
Isomers, relative energy Er (in eV), binding energies Eb (in eV), dissociation energy (in eV), HOMO-LUMO gap (in eV) for H2M3-Y (M = Li and Na and Y = AlF4, BeF3 and NO3) clusters.
To investigate the electronic interactions within the H2M3-AlF4 clusters, we computed NPA charges. The analysis reveals that all isomeric forms carry a net charge of approximately +0.93e at H2Li3 and +0.97e at H2Na3, while the AlF4⁻ unit holds −0.95e, indicating significant charge transfer and confirming the ionic nature of bonding. This result aligns with experimental observations, where AlF4
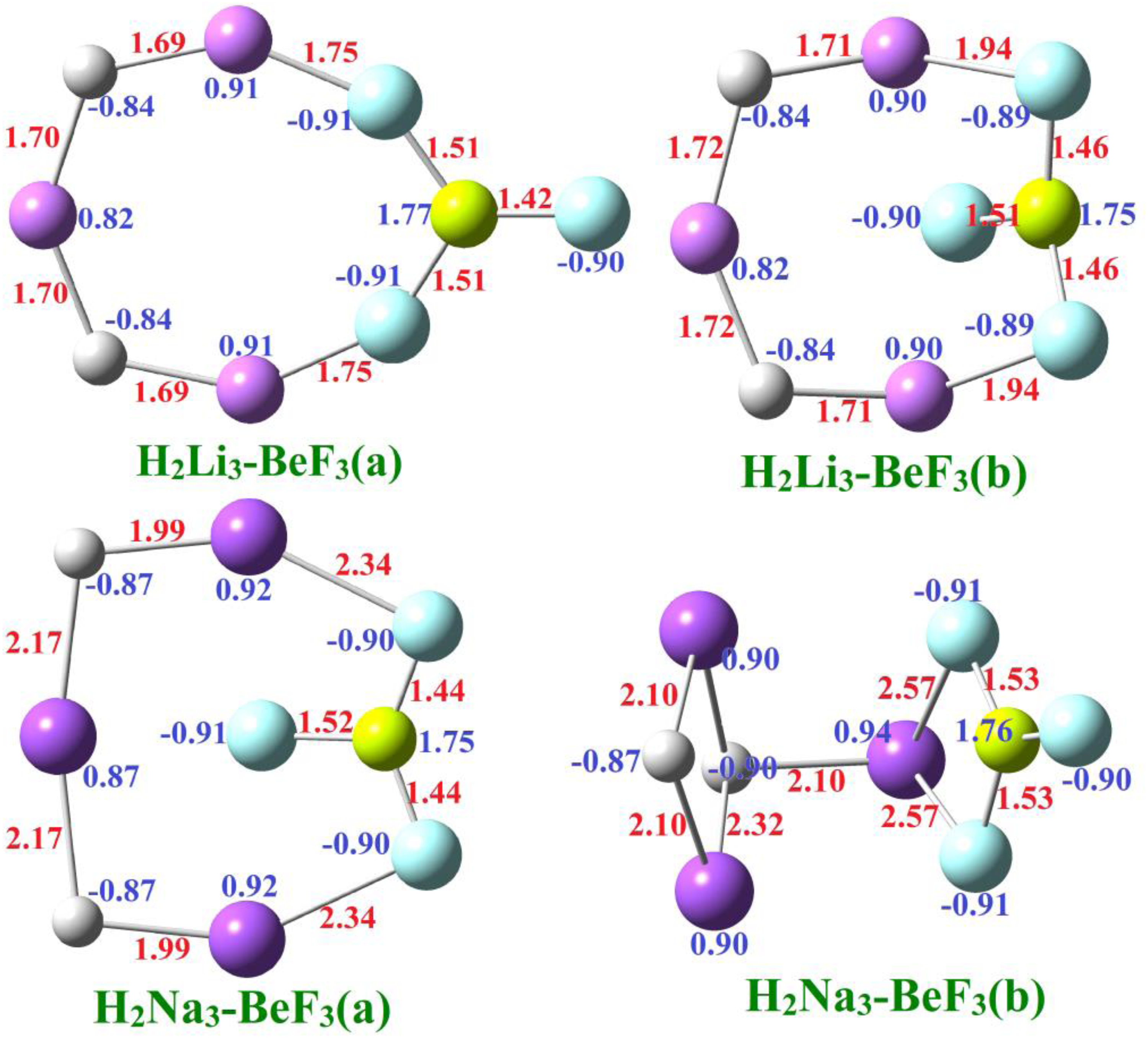
Optimized geometries of isomers H2M3-BeF3 (M = Li and Na) with selected bond lengths in Å and net charges at the MP2/aug-cc-pVTZ method.
The Be-F bond length is 1.42 Å, whereas a slight stretch in the Be-F bond length (1.51 Å) is noticed when the F-atom interacts with the Li- atom of the H2Li3. Similarly, in H2Na3-BeF3(b), the Be-F bond length is 1.52 Å, and the contraction in the bond Be-F is noticed in the interaction of BeF3 with the H2Na3. This behavior occurs because Li, being smaller and more polarizing than Na, pulls more electron density toward F, causing greater Be–F bond elongation. In contrast, larger Na interacts more loosely, leading to less Be–F bond distortion or slight contraction.
We further examined the superhalogen anions BeF3 and NO3, both of which have received extensive experimental validation in gas-phase and condensed-phase studies. In particular, high-resolution photoelectron spectroscopy (PES) of NO3 reveals distinct vertical detachment features around 4.66 eV, confirming its strong electron-accepting nature. 25
Additionally, vibrational spectroscopic investigations identify the antisymmetric stretching mode (ν₃) of NO3 near 1349 cm−1, supporting its structural rigidity. 26 While direct PES data for BeF3 are limited, gas-phase thermochemical studies on related clusters such as Be2F5 report significant clustering enthalpies (∼290 ± 10 kJ/mol), providing indirect yet compelling evidence of its robust ionic bonding behavior. 27 These experimental insights align well with our computed binding energies and electronic properties of the corresponding supersalts.
To identify the most stable structures, a wide range of initial geometries (including different orientations and coordination modes between the superalkali and superhalogen units) was constructed and optimized using the same computational scheme. During the optimization process, several trial structures converged to higher-energy configurations, showed significant structural rearrangements, or exhibited imaginary frequencies, and were therefore discarded. Only structures that converged without imaginary frequencies were considered true minima and are reported here. The optimized structures of H2M3-NO3 (M = Li and Na) are positioned in Figure 3. The oxygen center of the NO3 moiety establishes interactions with two Li atoms in H2Li3 and two Na atoms in H2Na3, respectively.
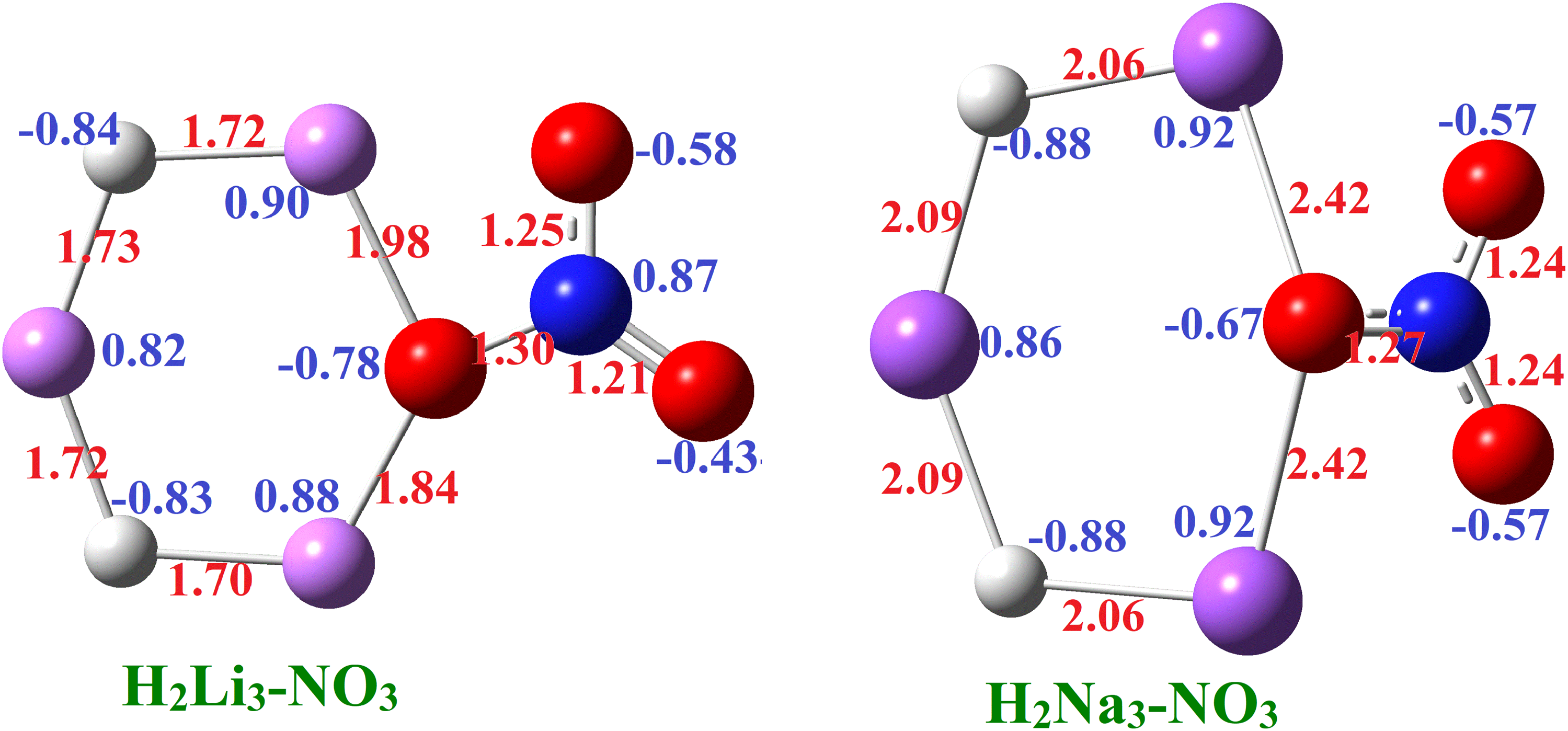
Optimized geometries of isomers H2M3-NO3 (M = Li and Na) with selected bond lengths in Å and net charges at the MP2/aug-cc-pVTZ method.
The net charge on the H2Li3 and NO3 is +0.93e and −0.92e, respectively. Thus, the calculated charges are comparable to the charges of H2M3-AlF4 and H2M3-BeF3 (M = Li and Na). The unequal O–Li bond lengths in H2Li3-NO3 arise due to differences in charge distribution, influencing Li–O interactions. Also, the estimated net charge on the H2Na3 and NO3 is +0.86e and −0.85e, respectively. The N-O bond length ranges from 1.30 Å to 1.21 Å in H2Li3-NO3, and in H2Na3-NO3, it ranges from 1.27 Å to 1.24 Å. Thus, no appreciable changes were noticed in the bond length on changing the superalkalis.
Stability and electronic properties
The stability and electronic properties of the resulting supersalts are analyzed using binding energy, dissociation energy, and the HOMO-LUMO gap, respectively, to highlight their distinct characteristics. A higher binding energy (Eb) indicates a stronger interaction and greater stability of the complex. Among all systems, H2Li3-AlF4 (Eb = 11.35 eV, Ed = 5.87 eV) possesses the highest binding and dissociation energies, indicating a strong interaction within the complex. The systems H2Li3-BeF3 and H2Na3-BeF3 exhibit intermediate stability between AlF4 and NO3 complexes. The observed stability trend (AlF4 > BeF3 > NO3) arises from differences in coordination environment, ligand electronegativity, and the extent of charge delocalization in the superhalogen units. The AlF4 anion possesses a highly symmetric tetrahedral geometry with four strongly electronegative fluorine atoms surrounding the central Al atom, thereby facilitating effective charge delocalization and strong electrostatic interactions with the superalkali fragment. In contrast, BeF3 contains only three fluorine atoms, resulting in slightly reduced charge delocalization and weaker electrostatic stabilization compared to AlF4. On the other hand, the planar NO3 distributes negative charge over the less electronegative oxygen atoms, which interact less strongly with the Li/Na centers, leading to comparatively weaker binding.
The H2Li3-BeF3 system has higher binding energy (Eb = 10.66 eV) and dissociation energy (Ed = 6.08 eV) than H2Na3-BeF3 (Eb = 9.34 eV, Ed = 4.86 eV), reflecting stronger Li interactions. The energy gap (Egap) is highest in H2Li3-BeF3 (10.33 eV), while Na-based systems exhibit lower Egap, indicating relatively higher electronic softness. Overall, BeF3 complexes are more stable than NO3 but less stable than AlF4, with Li-based systems consistently stronger than Na-based ones. For NO3-based systems, H2Li3-NO3 (Eb = 7.58 eV, Ed = 6.20 eV) is more stable than H2Na3-NO3 (Eb = 6.60 eV, Ed = 5.33 eV). This trend is consistent across all three ligand types, indicating that Li⁺ forms stronger electrostatic interactions than Na⁺, in line with its smaller ionic radius and higher charge density.
The HOMO–LUMO gap (Egap) is an important parameter related to the electronic stability and chemical hardness of a system. A larger Egap generally indicates lower chemical reactivity and higher resistance to electronic excitation. To gain further insight into the electronic structure of the supersalt complexes, the frontier molecular orbitals were analyzed and are shown in Figure 4. Although both HOMO and LUMO were computed for evaluating the HOMO–LUMO energy gaps, only the HOMO orbitals are presented here, as they primarily reflect the electron-donating nature of the superalkali fragment. The LUMOs are largely delocalized over the superhalogen units and do not provide additional qualitative insight. The HOMO distribution for all investigated systems is predominantly localized on the superalkali fragment (H2Li3), reflecting the diffuse electron density associated with the superalkali unit. Meanwhile, the superhalogen moieties contribute mainly to deeper lying molecular orbitals. This orbital localization further supports the charge-separated nature of the supersalt complexes.
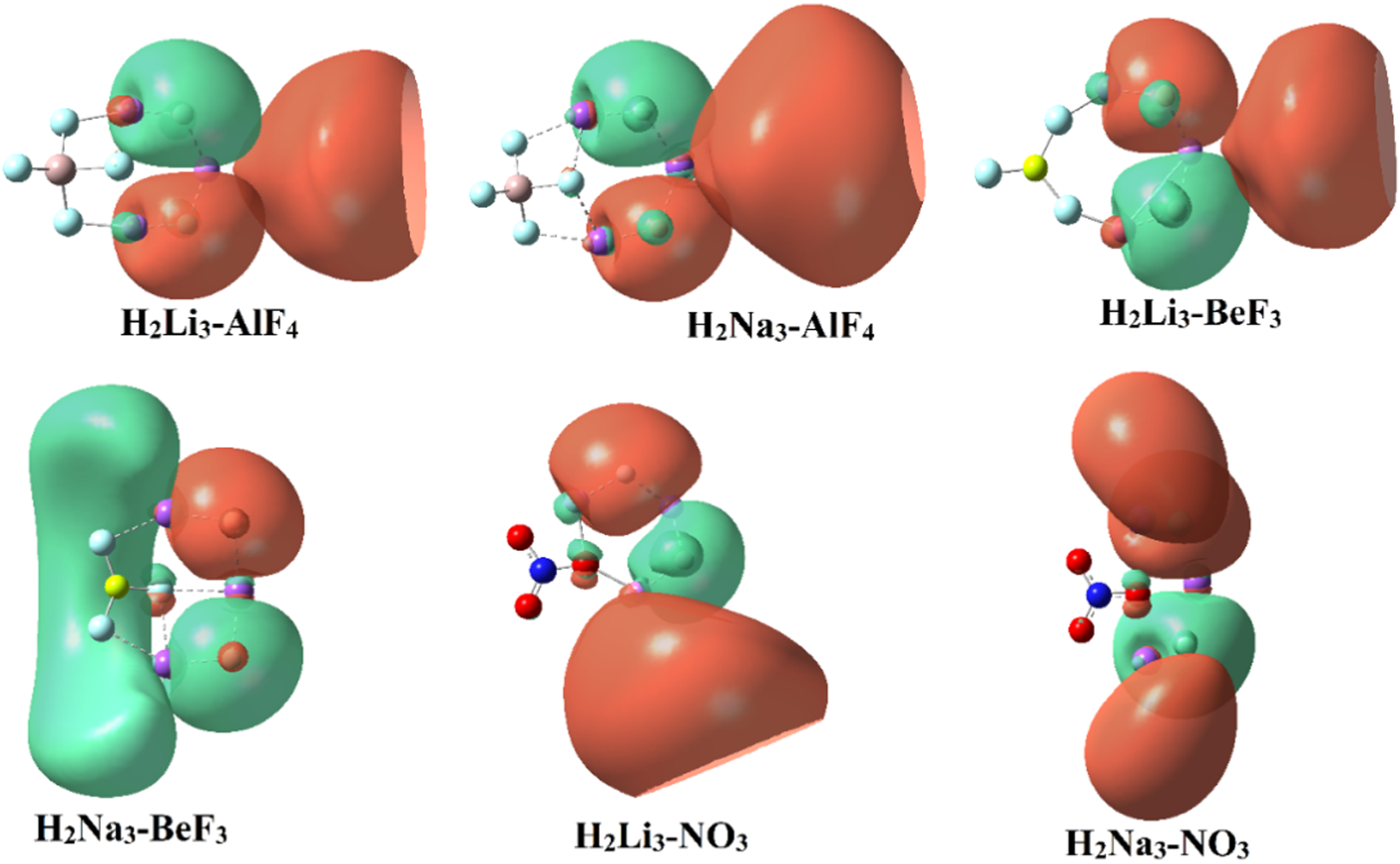
Frontier molecular orbitals (HOMO) of the optimized H2M3–Y supersalt complexes (M = Li, Na; Y = AlF4, BeF3, NO3) calculated at the MP2/aug-cc-pVTZ level. LUMOs are not shown as they exhibit similar delocalized features and do not provide additional insight.
Among the studied complexes, H2Li3-BeF3 exhibits the highest Egap (10.33 eV), suggesting improved electronic stability. Similarly, H2Li3-AlF4 (10.07 eV) and H2Li3-NO3 (9.99 eV) also possess relatively high energy gaps, highlighting the enhanced stability of Li-based systems. In comparison, the Na-based counterparts demonstrate slightly lower Egap values, with H2Na3-AlF4 (8.87 eV), H2Na3-NO3 (8.63 eV), and H2Na3-BeF3 (8.52 eV), indicating relatively lower stability. This trend indicates that replacing Li with Na results in a reduction in the electronic stability of the complexes. Additionally, the variation in Egap among different superhalogen clusters (AlF4, BeF3, NO3) highlights the influence of ligand electronegativity and structural effects on the electronic properties of these systems.
These trends indicate that Li-based systems not only exhibit stronger electrostatic and covalent interactions but also show larger HOMO–LUMO gaps. These observations are based on isolated molecular clusters and therefore reflect relative electronic stability at the molecular level. The nature of the superhalogen ligand significantly affects the complex stability, in the order AlF4 > BeF3 > NO3, which can be attributed to their inherent electron affinities and structural rigidity.
QTAIM analysis
The bonding characteristics of the supersalt complexes formed by superalkalis (H2Li3, H2Na3) and superhalogens (AlF4, BeF3, NO3) can be understood through various topological parameters, including electron density (ρ), Laplacian of electron density (∇2ρ), kinetic energy density (G), potential energy density (V), the energy ratio -G/V, and total energy density (H). In addition, the electron localization function (ELF) was computed to visualize the distribution of localized electrons within the complexes. The corresponding ELF maps are provided in the Supporting Information (Figure S12). Table 2 presents the analysis of several systems involving different interactions, where key quantities such as electron density (ρ), Laplacian of the electron density (∇2ρ), Lagrangian kinetic energy (G), potential energy density (V), and their corresponding ratios (-G/V) are reported for each interaction in H2M3-Y (M = Li and Na and Y = AlF4, BeF3 and NO3).
QTAIM parameters of neutral and cationic H2M3-Y (M = Li and Na and Y = AlF4, BeF3 and NO3) at MP2/aug-cc-pVTZ level. All values are given in a.u.
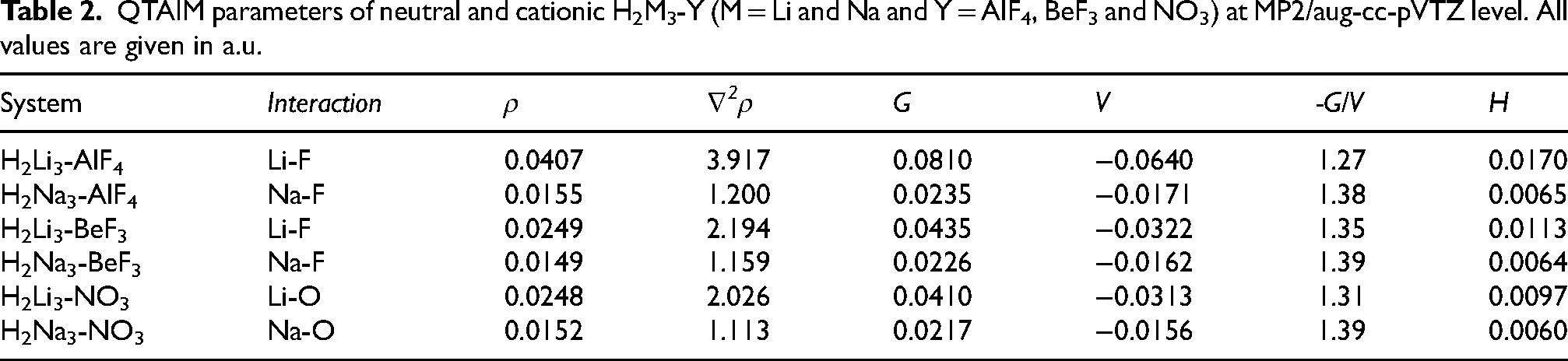
The electron density (ρ) at the bond critical point (BCP) is an important indicator of interaction strength, where higher values generally correspond to stronger bonding interactions. From Table 2, it is evident that Li-based systems exhibit higher electron density values compared to their Na-based counterparts. For example, H2Li3-AlF4 shows the highest ρ value (0.0407 a.u.), indicating relatively stronger interaction, whereas H2Na3-AlF4 shows the lowest value (0.0155 a.u.), suggesting comparatively weaker interaction. Similarly, H2Li3-BeF3 (0.0249 a.u.) and H2Li3-NO3 (Li–O interaction, 0.0248 a.u.) also display higher electron densities than the corresponding Na-based systems, such as H2Na3–BeF3 (0.0149 a.u.). This trend indicates that Li-containing supersalts possess stronger electron localization at the interaction region, leading to relatively stronger bonding interactions compared to the Na-based analogs.
The Laplacian of electron density (∇2ρ) provides important insight into the nature of bonding and the distribution of electron density within a system. In general, positive values of ∇2ρ correspond to closed-shell interactions such as ionic or van der Waals interactions, whereas negative values indicate covalent character. In all studied supersalt complexes, the Laplacian values are positive, confirming the predominance of ionic interactions between the superalkali and superhalogen units. A clear trend is observed: Li-based systems exhibit relatively higher ∇2ρ values than their Na-based counterparts, suggesting enhanced concentration of electron density in the interaction region. For instance, H2Li3-AlF4 and H2Li3-BeF3 show higher Laplacian values of 3.917 and 2.194 a.u., respectively, compared with H2Na3-AlF4 (1.200 a.u.) and H2Na3-BeF3 (1.159 a.u.). Among all systems, H2Li3-AlF4 exhibits the highest ∇2ρ value, further supporting strong electrostatic interactions in this complex.
The kinetic energy density (G) and potential energy density (V) provide important insights into the nature and stability of the bonding interactions. The ratio −G/V, which is greater than 1 for all complexes, indicates closed-shell interactions and confirms the predominantly ionic character of the supersalt systems. The −G/V ratio is highest for H2Na3-BeF3 (1.39) and H2Na3-NO3 (1.39), suggesting a relatively stronger ionic interaction character, whereas H2Li3-AlF4 (1.27) shows a slightly lower value, implying stronger electrostatic interactions. In general, Li-based systems exhibit higher kinetic energy density and more negative potential energy density values, indicating relatively stronger interactions. Although Na-based systems display slightly larger −G/V ratios, their overall electron densities and binding energies remain lower than those of the corresponding Li-based systems. Consequently, Li-based supersalts are energetically more stable despite the somewhat higher ionic character observed in Na-containing complexes.
The total energy density (H), defined as H = G + V, determines the overall stability of interactions. Positive values of H, as seen in all complexes, indicate weakly bound interactions, reinforcing the ionic nature of bonding. Similar closed-shell interaction features and electron density characteristics have also been reported for supramolecular systems stabilized through excess-electron interactions, supporting the general relevance of such bonding behavior in charge-separated molecular assemblies. 15
The interaction between the H2M3 superalkali unit and the superhalogen moiety primarily occurs through Li/Na–F contacts in fluorine-containing superhalogens (AlF4 and BeF3), whereas Li/Na–O interactions dominate in nitrate-based systems. This difference arises because, in NO3−, the negative charge is primarily localized over oxygen atoms, making them the preferred interaction sites. The QTAIM analysis further confirms that the bond critical points are located between Li/Na and F atoms for AlF4 and BeF3 systems, and between Li/Na and O atoms for NO3 systems, consistent with the structural description of oxygen-mediated interactions observed in nitrate-based complexes. Overall, the H2Li3 complexes exhibit stronger interactions than their H2Na3 counterparts due to higher electron density and lower −G/V ratios, thereby rendering lithium-based supersalts more stable. Among all, H2Li3-AlF4 shows the strongest interaction, making it the most stable supersalt, while H2Na3-AlF4 has the weakest bonding. These findings confirm that superalkali-superhalogen interactions lead to highly ionic supersalt formations, with bond strength following the trend AlF4 > BeF3 > NO3, and lithium-based complexes being more stable than sodium-based ones.
Conclusions
In this study, a series of typical supersalts with the general formula H2M3–Y (where M = Li or Na, and Y = AlF4, BeF3, or NO3) was investigated. These systems represent novel compounds formed by combining superalkalis with superhalogens. All calculations were performed at the MP2/aug-cc-pVTZ level of theory. The structural properties, particularly bond lengths, were calculated for each possible configuration of these supersalts. Their stability was evaluated using two key energetic parameters: binding energy (Eb) and dissociation energy (Ed). The results indicate that these supersalts experience significant stabilization due to charge transfer from the superalkali unit to the superhalogen fragment.
To probe the electronic properties, the HOMO–LUMO energy gap (Egap) was calculated. Among the studied complexes, H2Li3–BeF3 exhibits the highest Egap value of 10.33 eV, suggesting strong electronic stability. Analysis of net atomic charges revealed effective charge separation between H2M3 and the superhalogen moieties, supporting their ionic character. This ionic nature of bonding was further validated through QTAIM analysis, which confirmed significant electron localization between interacting centers.
Comparative results demonstrate that Li-based supersalts consistently show greater stability, stronger intermolecular interactions, and wider HOMO–LUMO gaps than their Na-based analogs. Additionally, among the superhalogens examined, AlF4 exhibits the highest binding energy, followed by BeF3 and NO3, indicating that the choice of superhalogen significantly influences both the structural integrity and electronic behavior of the resulting complexes. Overall, these findings highlight the critical role of the central metal atom (Li vs. Na) and the nature of the superhalogen in determining the stability, bonding, and electronic properties of supersalts. This work provides theoretical insight into charge-transfer-driven molecular systems.
Supplemental Material
sj-docx-1-mgc-10.1177_10241221261456942 - Supplemental material for Novel supersalts formed by interaction of H2M3 superalkalis (M = Li and Na) with AlF4, BeF3 and NO3 superhalogens
Supplemental material, sj-docx-1-mgc-10.1177_10241221261456942 for Novel supersalts formed by interaction of H2M3 superalkalis (M = Li and Na) with AlF4, BeF3 and NO3 superhalogens by Harshita Srivastava and Ambrish Kumar Srivastava in Main Group Chemistry
Footnotes
Ethical considerations and informed consent
Not applicable
Author contributions
HS: literature survey, calculations, data collection, and writing draft. AKS: conceptualization, supervision, editing, and finalizing the draft.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr Ambrish Kumar Srivastava acknowledges the funding received from the Science & Engineering Research Board (SERB), New Delhi, through the State University Research Excellence (SURE) scheme (SUR/2022/002035).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Data associated with this article will be available upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.