
Abstract
In this quantum chemistry study, the B2H6…B3S3H3 interaction was illustrated at M062X/6-311G(d,p) level of theory. Substitution of hydrogens of borthiin with F, Cl, CN and Me substituents was clarified. Energy decomposition analysis (EDA) was considered to exploring of the nature of the B2H6…B3S3X3 interactions. The interacting sites of the two fragments were reflected with molecular electrostatic potential (MEP) surfaces. Substituent effect on the polarity, electronic spatial extent and HOMO-LUMO gap of the B2H6…B3S3X3 complexes were stated. Quantum theory of atoms in molecules (QTAIM) analysis was considered to determination of bond critical points (BCP) between B2H6 and B3S3X3 molecules. The electron transfer between two molecules was studied with charge decomposition analysis (CDA). Besides, interactions between B2H6 and B3S3X3 molecules were exposed with interacting quantum atoms (IQA) approach.
Introduction
Inorganic aromatic molecules have been interesting molecules in the inorganic chemistry. Therefore, attentions of inorganic chemists exited to preparation and characterization of inorganic aromatic rings. Borthiin (B3S3H3) molecule is one of this family compounds. Preparations and characterizations of this molecule have been excited much attentions. In a study synthesis of B3S3(NMe2)3 molecule has been published. 1 Quantum chemistry tools have been illustrated structure and properties of borthiin molecule.2–6
Energetics, structural, and spectral parameters have been considered to illustration of hydrogen bonding (H-bonding) interaction.7–16 A “Hydrogen Bridge” has been used to discrete all of the non-bonded interactions in which hydrogen atom bridge between any two atoms. 17 Numerous boron compounds contain bridge hydrogen atoms. For model, boron hydrides expose unique bonding characters and have involved many researches due to their exclusive chemical properties.18–22 Diborane (B2H6) molecule has been selected as a typical compound for the consideration of an interaction between n-B18H22 and benzene in the molecular crystal. 23 In this paper, the Hbridge atom of n-B18H22 straight interacts with the center of the C6H6 cycle.
The diborane … π-systems interaction has been motivated various investigations.24,25 For example, substitution effects in the interaction of diborane and borazine have been illustrated. 26 In other study, a considered hypothetical reaction has been explored electronic structure of the Hbridge bond in diborane. 27 A computational research on substituent effect of terminal hydrogens of B2H6 with F atoms on the Borazanaphthalene…B2H6 interaction has been published. 28
In this quantum chemistry research, we attracted to understanding the interaction of B2H6 and borthiin. The substitution effect of hydrogens of borthiin on the structural, and electronic properties of theses systems was explored.
Computational methods
Optimization of the studied molecules was provided with hybrid functional of Truhlar and Zhao (M06-2X)functional, 29 6-311G(d,p) basis set30–33 and the software package of Gaussian 09. 34 Computations of harmonic vibrational frequencies were indicated no imaginary frequency for optimized molecules.
Energy decomposition analysis (EDA) was worked to understanding of interaction between the thiophene and nanoclusters with Multiwfn 3.8 software package.35,36 The interaction energy (ΔEint) between the B2H6 and B3S3X3 molecules was valued as:
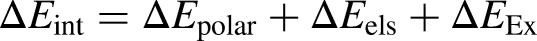
Molecular electrostatic potential (MEP) visualized by the GaussView-5.0 program. Atomic charges Atomic dipole corrected Hirshfeld atomic charge (ADCH) analysis, 37 and charge decomposition analysis (CDA) 38 were accomplished using Multiwfn 3.8 software package.35,39
Results of quantum theory of atoms in molecules (QTAIM) analysis and interacting quantum atoms (IQA) were provided with the AIMAll software package. 40 The “Promega” basin integration method using fine interatomic surface mesh was considered.
The IQA partitioning pattern employs the QTAIM description of atomic basins to estimate intra- and interatomic energy terms. Sum of these values is expressed as the total energy of the system:
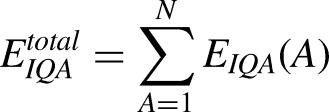
Each atomic energy can be extended as a sum of intra- and interatomic contributions:
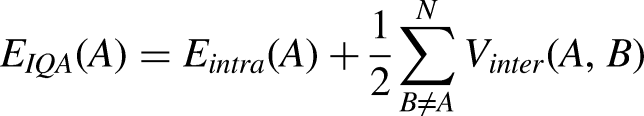
The intra-atomic term, Eintra (A), is the sum of the (intra-atomic) kinetic energy, T(A), and electron-electron, Vee (A), and electron-nucleus Ven (A) potential energies.
The interatomic contributions are stated by:
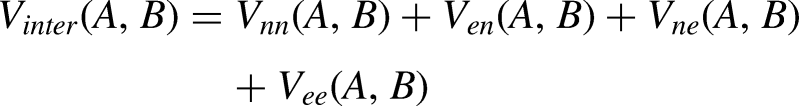
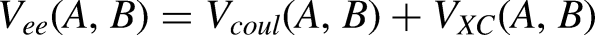
All Coulombic terms can be gathered together into the classical potential term Vcl(A,B).
Then, the interatomic term is stated as:
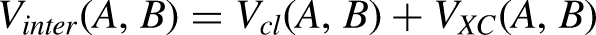
Results and discussion
Figure 1 presents the optimized geometries of the B2H6…B3S3X3 (X = H, F, Cl, CN, Me) systems at the M062X/6-311G(d,p) level of theory.
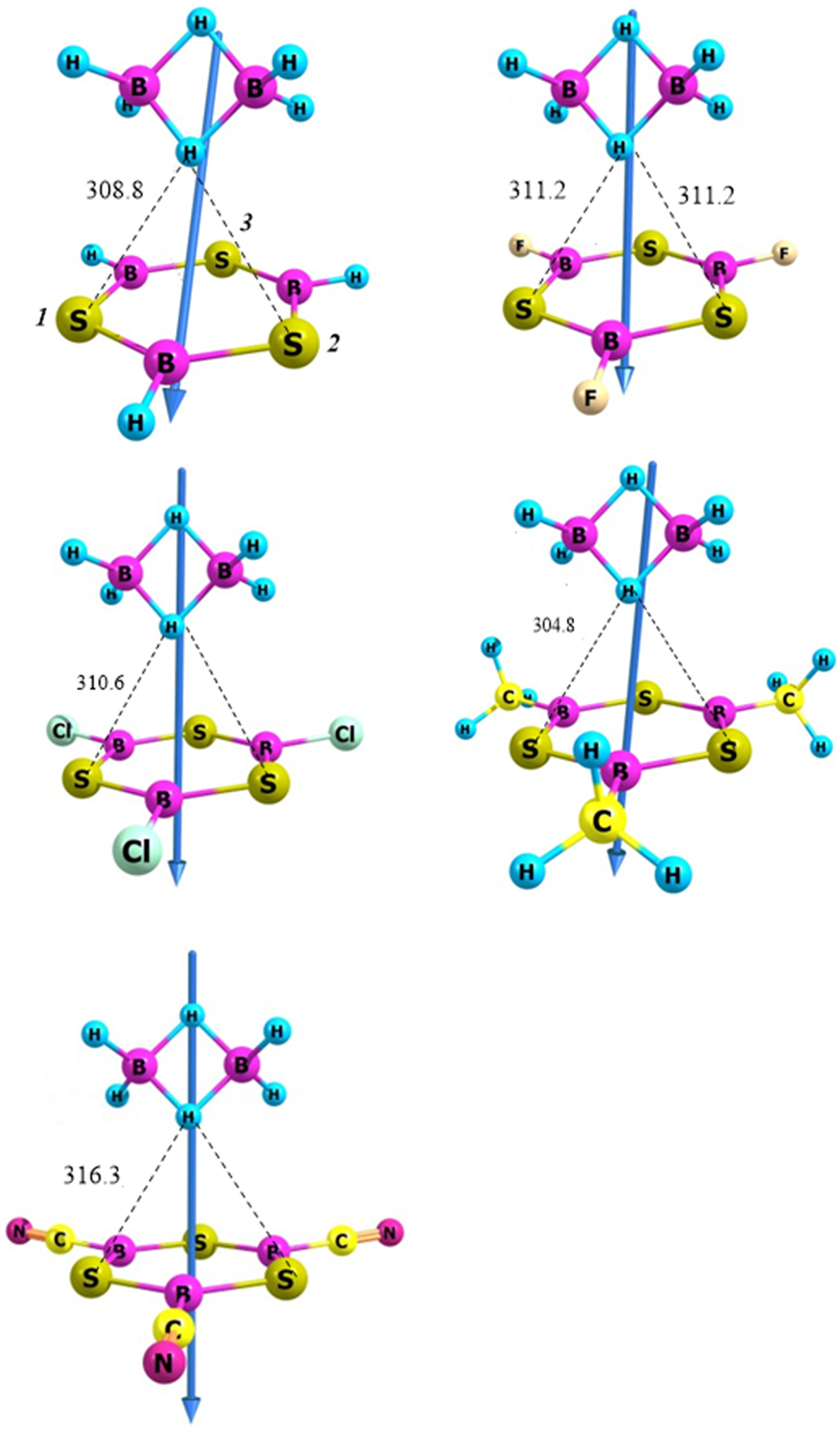
The optimized geometries and direction of dipole moment vector of the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at the M062X/6-311G(d,p) level of theory.
Energy decomposition analysis (EDA)
Energy decomposition analysis (EDA) is useful tool to clarification the B2H6 …B3S3X3 (X = H, F, Cl, CN, Me) interactions. The calculated interaction energy data (ΔEint) are documented in Table 1. It can be inferred stronger interaction arises with replacing of hydrogen atoms with substituents. The interaction strength decreases as Me > F > Cl > CN > H. Figure 2 indicates diagram of −ΔEint variations of the B3S3X3…B2H6 systems.
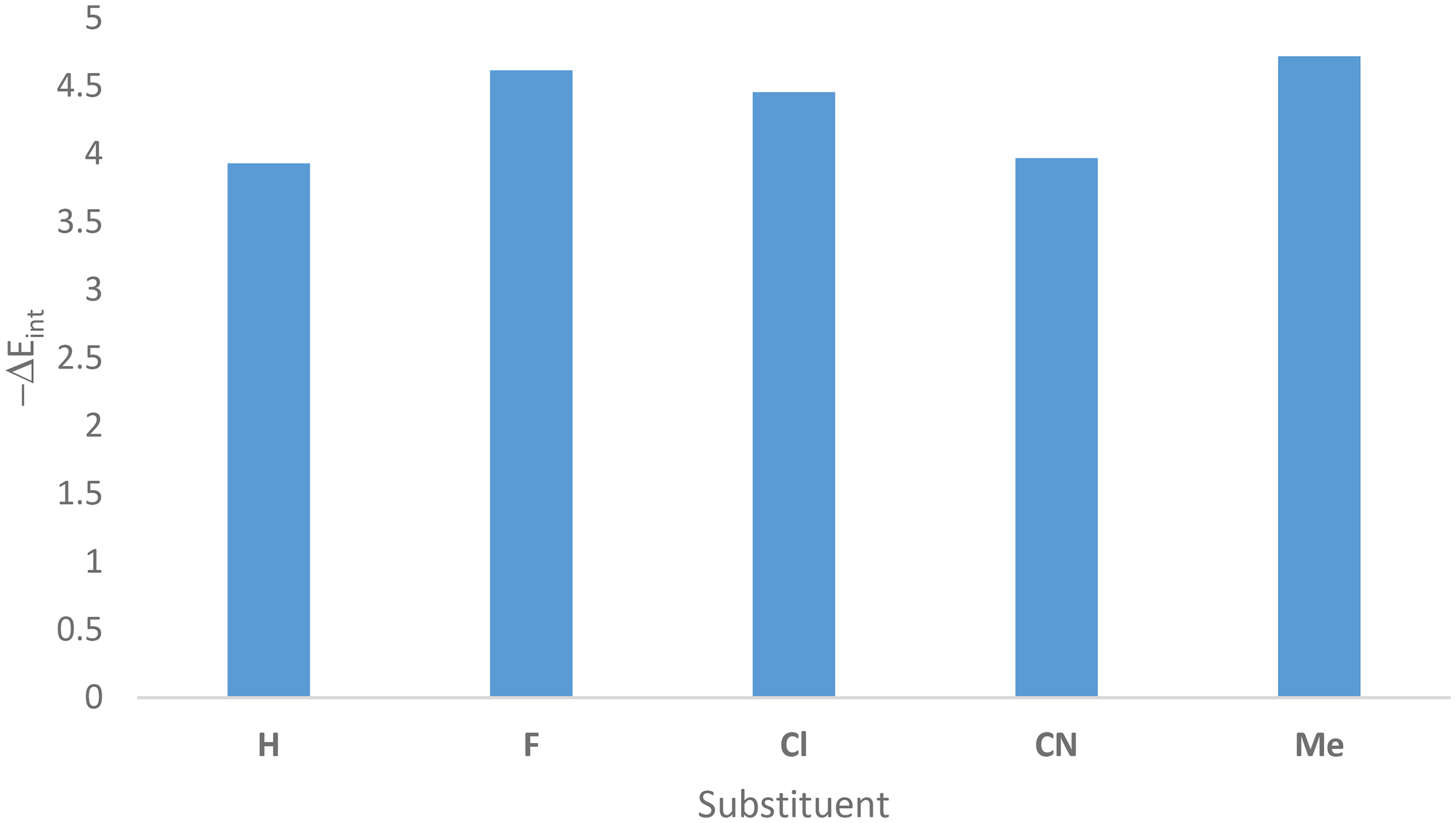
Diagram of −ΔEint variations of the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at the M062X/6-311G(d,p) level of theory.
EDA results of the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at M062X/6-311G(d,p) level of theory (in kcal/mol).
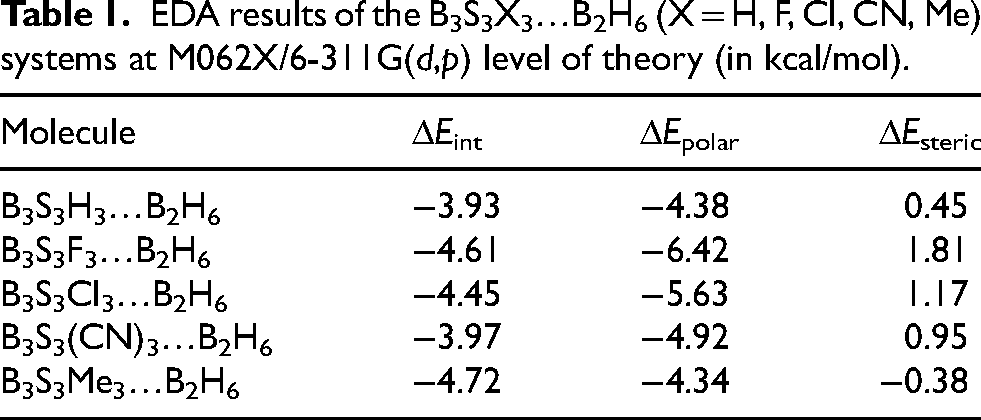
The major reason for this trend is attributed to in the nature of the interaction. B2H6 and B₃S₃X₃ molecules are classic Lewis acid and Lewis base, respectively. The strength of the Lewis acid-base interaction is straight related to electron donating ability of the base. Me group includes electron-donating ability. It makes the B₃S₃X₃ ring more electron-rich and highly attractive to the electron-deficient B2H6.
On the other hand, halogens are highly electronegative and would seem to be electron-withdrawing. But, they also include lone pairs of electrons that can be donated into the ring through a resonance effect. For fluorine and chlorine, this lone pair donation (resonance) is noteworthy and partially frustrates their strong inductive pull. This results in a net electron density that is moderately high, allowing for a decent interaction with B2H6.
Finally, the CN group is a powerful electron-withdrawing group. It pull out electron density through both a strong inductive effect (due to the electronegative nitrogen) and a strong resonance effect (via its π-bonds). This dual withdrawal leaves the B₃S₃X₃ ring electron-poor, creating it much less possible to interact strongly with B2H6.
The negative ΔEpolar values for all B2H6…B3S3X3 systems expose that these data stabilize complex systems. The most stability is found in the presence of fluorine substituent.
It can be provided positive ΔEsteric values for these systems, except X = Me. Accordingly, these values destabilize B2H6 …B3S3X3 systems. The most instability is seen in the presence of fluorine substituent. Hence, the main driving force of the B2H6 …B3S3X3 interactions is polarization or orbital-interaction stabilization.
Polarity
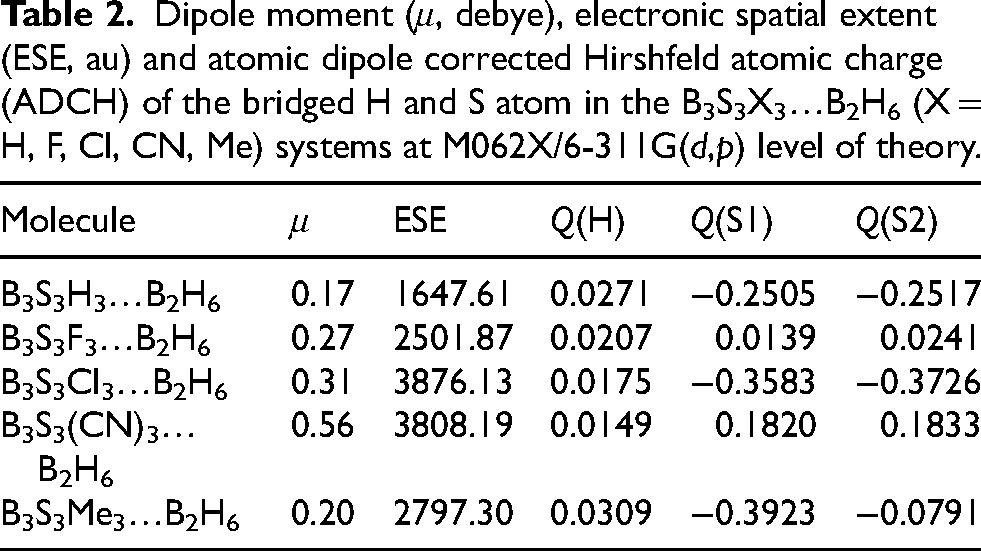
Computed dipole moment values of the B3S3H3−nX n …B2H6 (X = H, F, Cl, CN, Me; n = 0–3) systems are given in Table 2. These values show that replacing of hydrogen atoms of borthiin with F, Cl, Me, and CN substitutions increase polarity. These values reveal increase as H < F < Me < Cl < CN. Orientations of dipole moment vector of the molecules are presented in Figure 1. It can be found, most polarity is seen in the presence of CN group. This group is a long and rigid spacer that maximizes the distance between separated charges, yielding the largest dipole despite minimal electron donation. Cl substituent is large and polarizable, letting a smaller charge to be separated over a longer distance, increasing dipole moment. Me group donates the most density, but the charge is spread over a diffuse volume, limiting the dipole. Finally, F substituent gives small density, so charge separation is modest.
Dipole moment (μ, debye), electronic spatial extent (ESE, au) and atomic dipole corrected Hirshfeld atomic charge (ADCH) of the bridged H and S atom in the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at M062X/6-311G(d,p) level of theory.
Atomic dipole corrected Hirshfeld atomic charge (ADCH)
The calculated atomic dipole corrected Hirshfeld atomic charge (ADCH) on the bridged H and S atoms of B3S3H3−nX n …B2H6 systems are gathered in Table 2. Computed ADCH values of bridged hydrogen of B2H6 equals to 0.1167. on the other hand, calculated ADCH values of S atom of B3S3X3 molecules are −0.0446 (H), −0.0695 (F) −0.0338 (Cl), 0.0548 (CN) and −0.0960 (Me). The smaller charges of bridged H atom are found B3S3H3−nX n …B2H6 systems than in isolated B2H6 molecule (Figure 3). These charges decrease as X = Me > H > F > Cl > CN. It can be gotten from the outcomes that the ADCH values on the Hbridge atom that is involved in the interaction with the π-cloud reduce with respect to the isolated state in the presence of X = F, Cl and CN. In contrast, the ADCH value on the Hbridge atom enhances with respect to the isolated state in the presence of X = Me.
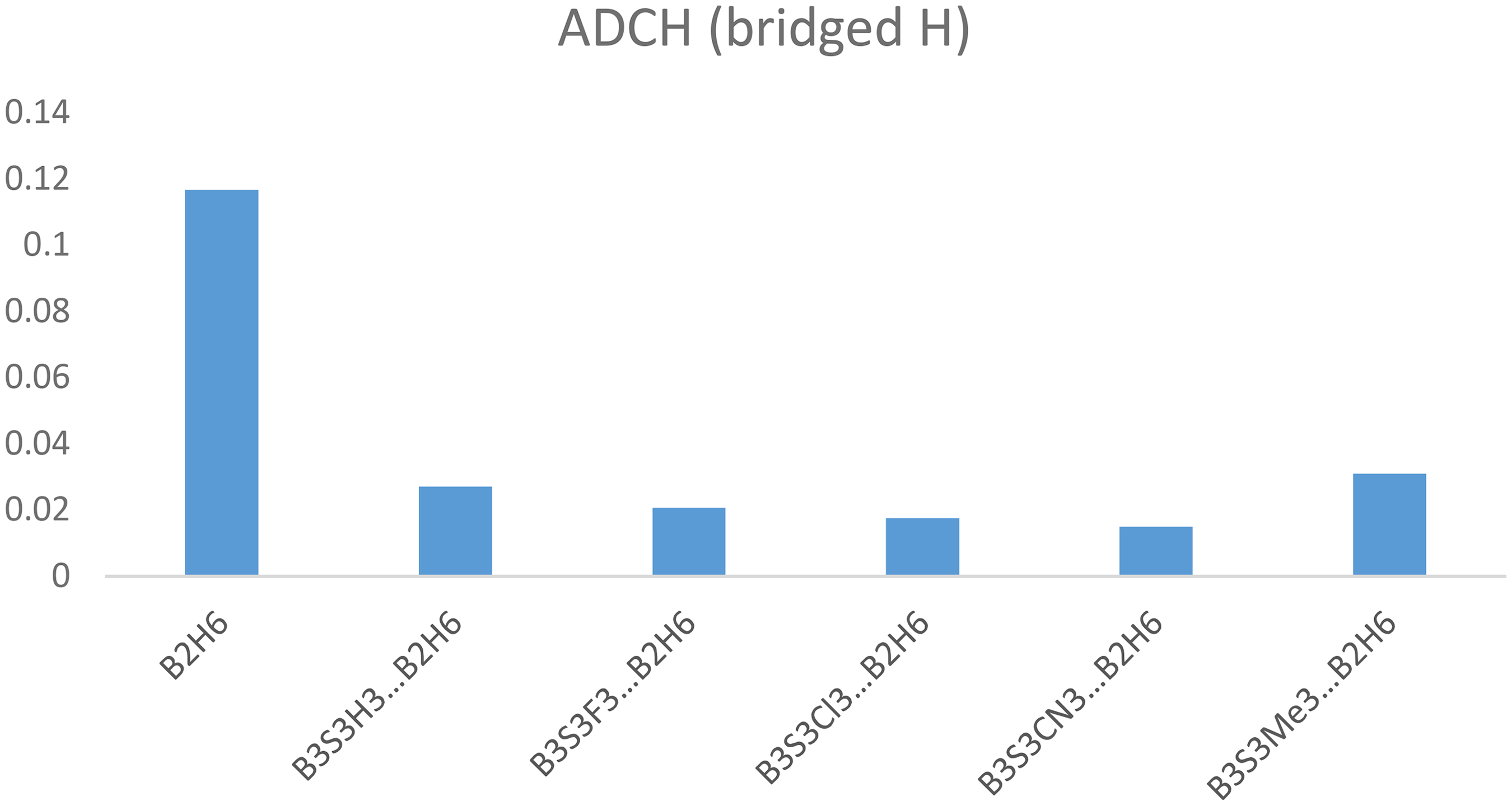
The calculated Hirshfeld atomic charges on the bridged H of B3S3H3−nX n …B2H6 in complexed and isolated B2H6 molecule in the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at the M062X/6-311G(d,p) level of theory.
Electronic spatial extent (ESE)
ESE is defined as the expectation value of the multiplied of electron density by the distance from a molecule mass center. More it is physical property of the electron density volume round the molecule. As the extent value enhances as electron cloud becomes more defused. Calculated ESE data of the B3S3H3−nX n …B2H6 systems are tabulated in Table 2.
Computed ESE values reveal replacing of hydrogen atoms of borthiin with F, Cl, Me, and CN substitutions increase ESE values. The ESE values increase as H < F < Me < CN < Cl. It must be noticed the ESE is dominated by two geometric factors: The intrinsic size of the substituent atom (Van der Waals radius) and the bond length between the ring and the substituent. The trend is a competition between how far the nuclei are spread out (geometry) and how tightly the electrons are held (electronegativity). It can be seen largest ESE in the presence of Cl substituent. It includes 3rd shell electrons (3s, 3p). These electrons are much farther from the nucleus than the 2nd shell electrons of F, C, or N. Besides, Cl substituent contains has lone pairs that are very “soft” and polarizable. Even though the B-Cl bond is strong, the electron cloud surrounding chlorine is obviously huge and diffuse. The slight negative charge that develops on Cl in the complex resides in these large, fluffy orbitals, causing in the largest ESE value. In linear CN group, N atom is small, the triple bond creates a rigid rod. This extends the substituent far away from the central borthiin ring. The π-electrons in the triple bond are also quite mobile. However, CN withdraws electron density, the distance of those electrons from the center of the complex is large, increasing ESE significantly. Me group has more significant volume than F substituent, however Me donates electrons (making the cloud denser), the physical presence of the carbon and three hydrogens pushes the electron cloud outward further than Fluorine's tight cloud. Fluorine electronegative substituent pulls electron density very close to its nucleus. Thus, while the nuclei are slightly further apart, the electrons are held tightly, limiting the ESE.
Frontier orbitals
Plots of frontier orbitals of the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems are presented in Figure 4. It can be deduced most contribution is belonged to B3S3X3 fragment.
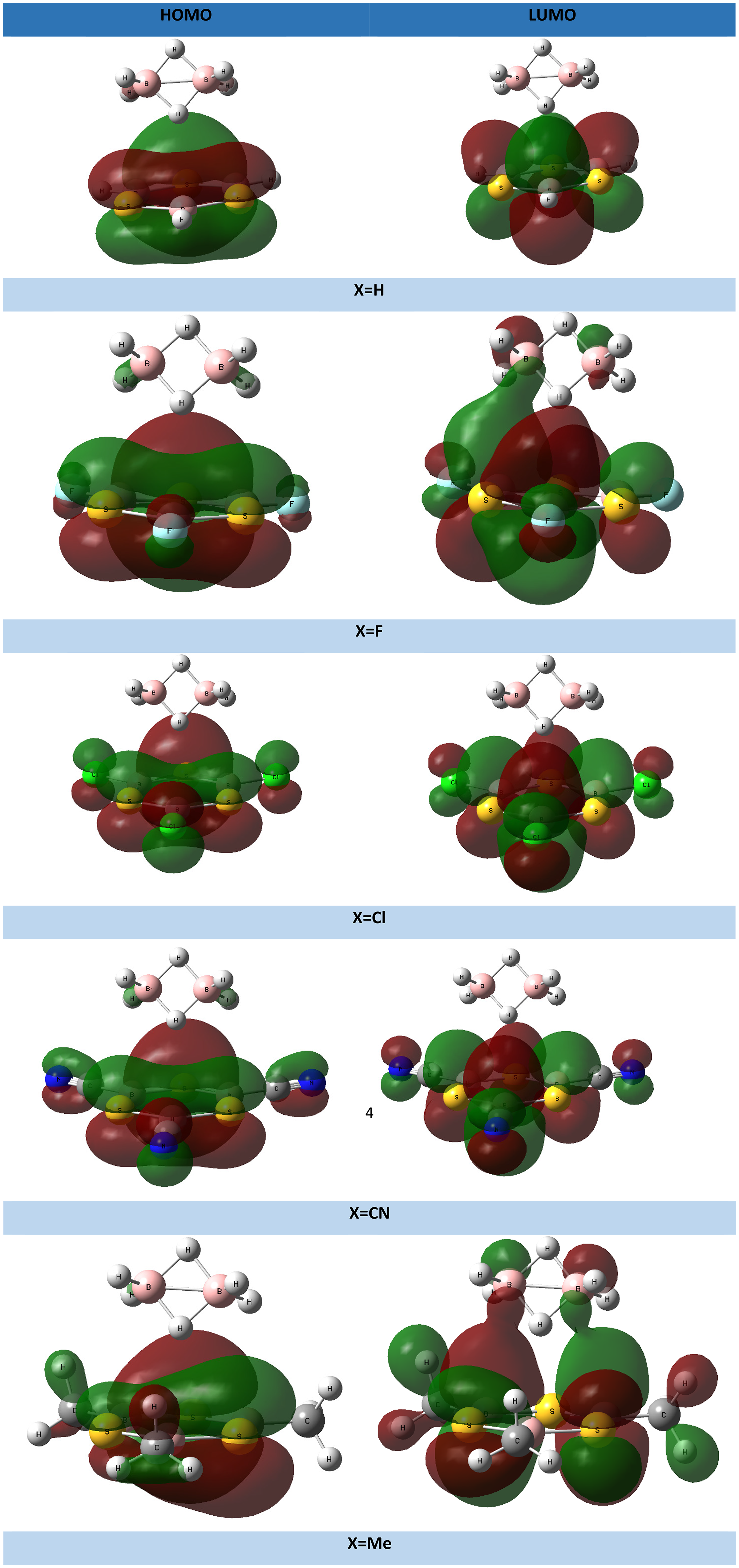
Plots of frontier orbitals of the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at the M062X/6-311G(d,p) level of theory.
Evaluated energy values of frontier orbitals of these systems are gathered in Table 3. These values show that the stability of HOMO decreases as X = CN > F > Cl > H > Me. On the other hand, the stability of LUMO decreases as X = CN > Cl > H > F > Me.
Frontier orbital energy and global indices of reactivity for the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at M062X/6-311G(d,p) level of theory (in eV).
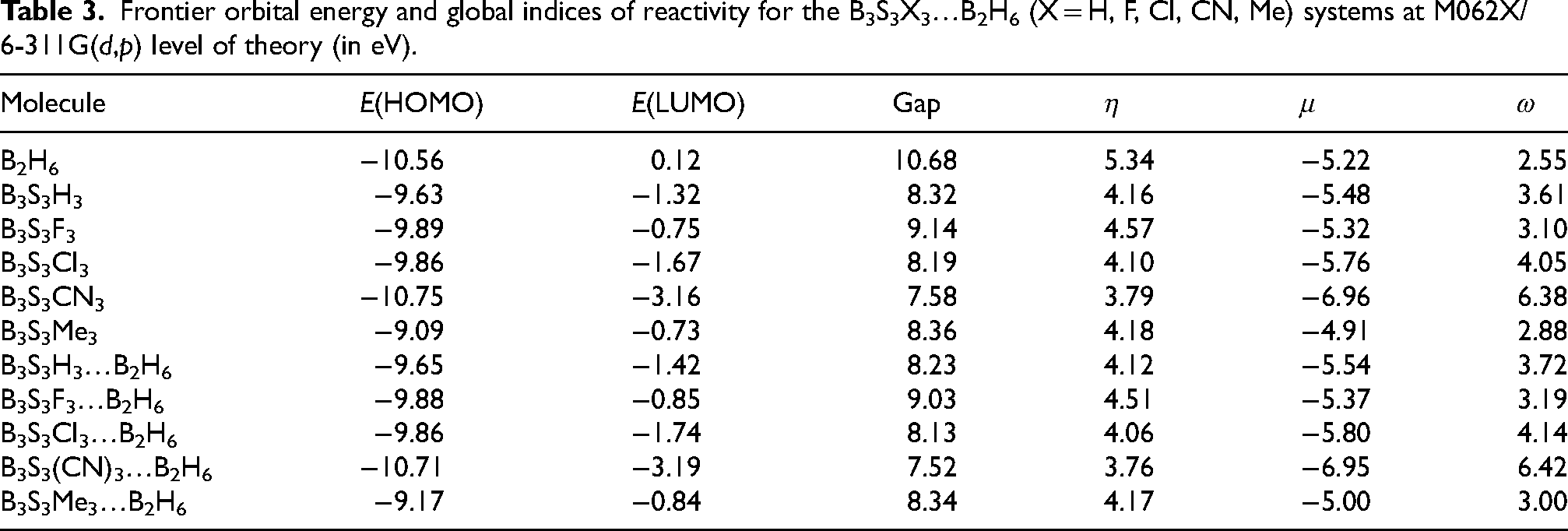
Computed HOMO-LUMO gap values of these systems are given in Table 3. These values decreases X = F > H > Me > Cl > CN. This trend in understandable in terms of substituent character. In the presence of F substituent, LUMO energy raises (via σ* mixing or lone-pair antibonding interaction) whereas E(HOMO) slightly lowering. Therefore, largest HOMO-LUMO gap can be observed. In the presence of Me group, E(HOMO) raises and E(LUMO) raises less respect to X = H. So, smaller HOMO-LUMO gap is found than X = H. Cl lowers E(HOMO) moderately and lowers E(LUMO) significantly (via d-orbital acceptance). Thus, smaller HOMO-LUMO can be seen. Finally, CN decreases E(HOMO) and decreases E(LUMO) even more (via π*- acceptance). Hence, smallest HOMO-LUMO gap can be created.
Molecular electrostatic potential (MEP)
Now, we used MEP surfaces for exploring of EDA results. The MEP surfaces of the B2H6 and B3S3H3 molecules can be help to exploring the binding sites in a rational approach for the B3S3H3…B2H6 complex. MEP surfaces of these molecules are shown in Figure 5. The MEP maps obviously exemplify negative areas S atom of B3S3H3 and a positive area around bridged hydrogen of B2H6 molecule. Therefore, interaction between two fragments is occurred between S atom of B3S3H3 and bridged hydrogen of B2H6 molecule.
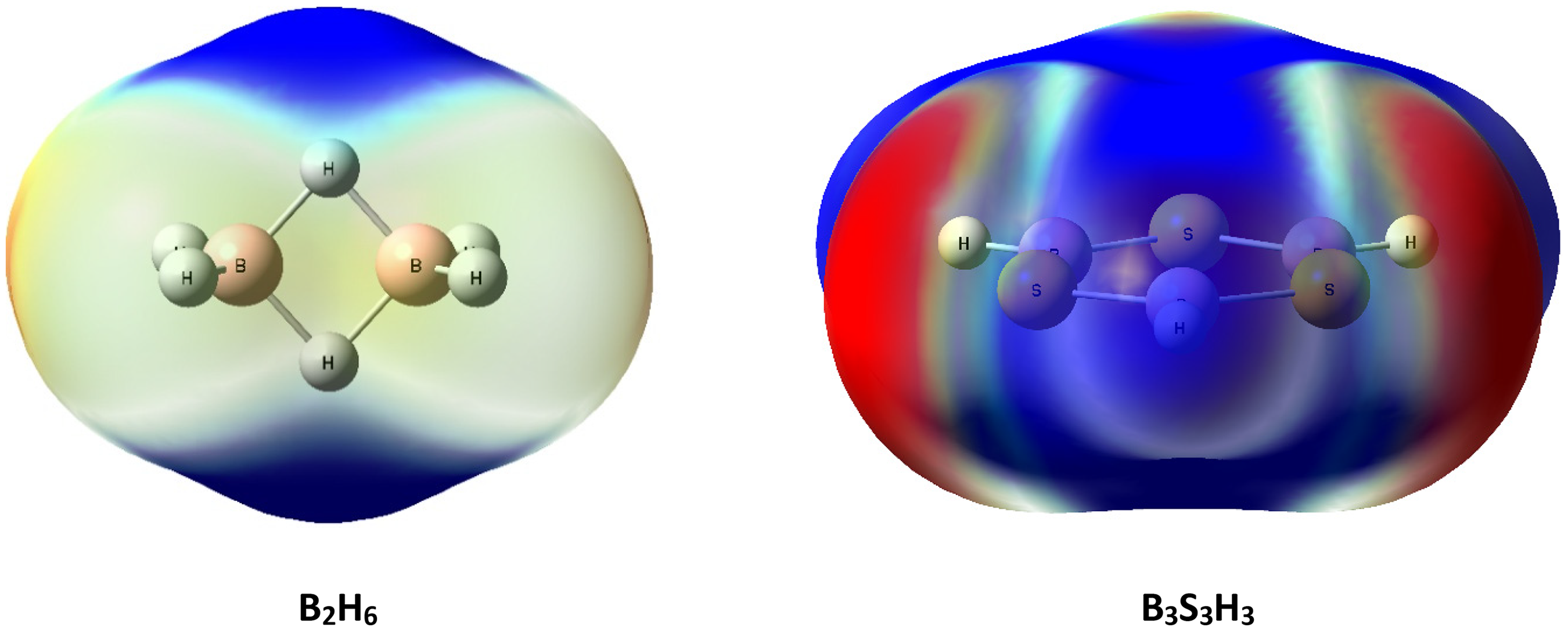
The MEP surfaces of the B2H6 and B3S3H3 molecules.
Charge decomposition analysis (CDA)
Now, we illustrated charge transfer between two fragments of B3S3X3…B2H6 systems using of decomposition analysis (CDA) theory. Table 4 lists the CDA results of these systems. The CDA outcomes express larger electrons donated from B3S3X3 fragment to the diborane molecule in the presence of X = H, F, Me. This variation can be attributed to better electron donating ability of these substituents. In contrast, greater electrons donated from diborane to B3S3X3 fragment in presence of X = Cl, CN. This variation is compatible with to better electron withdrawing ability of these substituents.
Charge decomposition analysis (CDA) results of the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at M062X/6-311G(d,p) level of theory.

d, b and r terms are the number of electrons donated from fragment B2H6 to fragment B3S3X3, the number of electrons back donated from fragment B3S3X3 to fragment B2H6, the number of electrons involved in repulsive polarization, respectively.
A minor (but non-negligible) value of
QTAIM
Now, we provided worth idea about the bond critical points (BCP) of S…H of B2H6…B3S3X3 molecules with QTAIM computations. Two bond critical points are accepted at S1…H and S2…H bonds. Computed ρBCP(S…H) values are recorded in Table 5. It can be observed the ρBCP(S…H) values decrease as X = Me > H > Cl > F > CN. This trend explains with variation of S…H distances. S…H distances decrease with increasing of ρBCP(S…H) values.
S…H bond distance (in Å) and QTAIM results of the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at M062X/6-311G(d,p) level of theory.
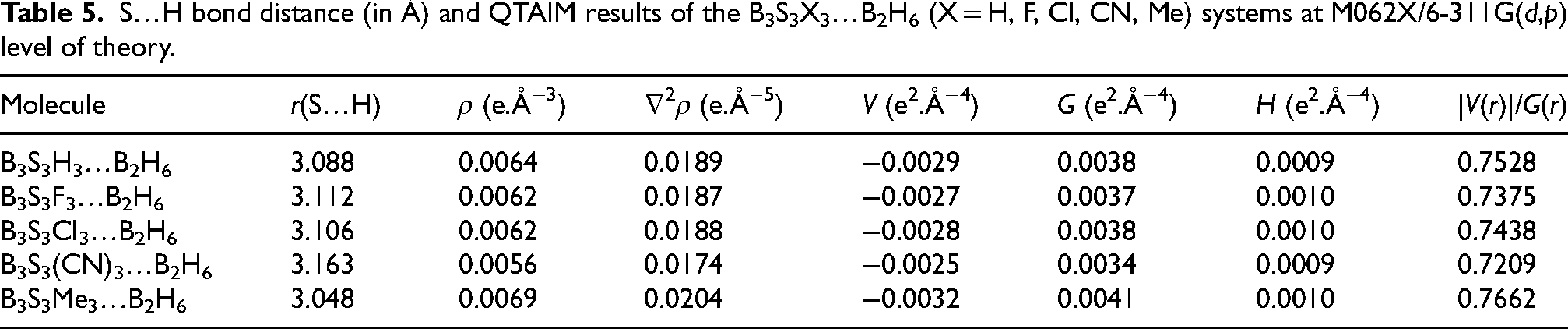
Calculated ∇2ρBCP(S…H) values are recorded in Table 5. These positive values are well-matched with the S…H closed-shell interactions.
Calculated density of total electron energy (H, S…H) values are recorded in Table 5. The positive H values are reflected as a van der Waals interaction indicator.
Evaluated |V(r)|/G(r) ratio values are summarized in Table 5. |V(r)|/G(r) < 1 values are characteristic of van der Waals interaction. 41
Interacting quantum atoms (IQA) approach
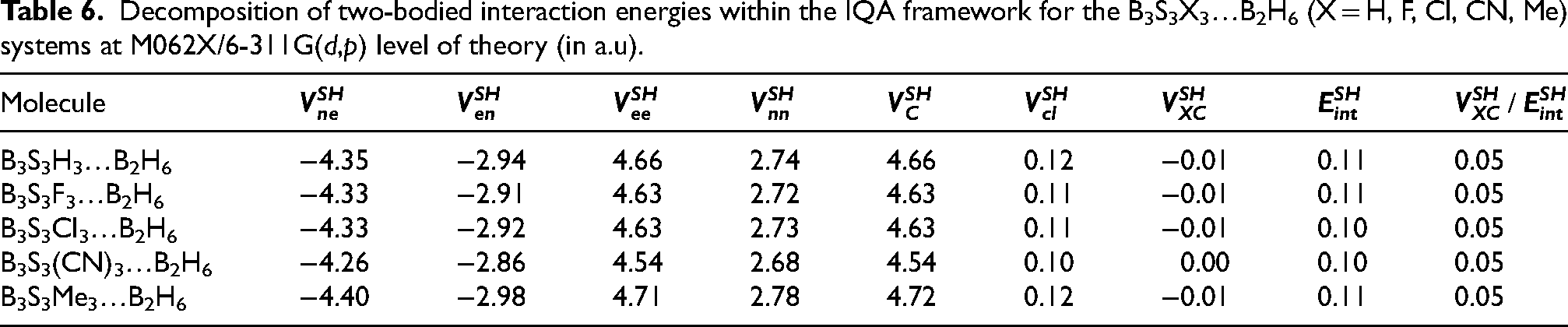
Interacting quantum atoms (IQA) method is valuable tool to exploring of metal-ligand interactions mechanisms in the B2H6…B3S3X3 systems was explored with the atomic energy partitioning method, namely.42–45 The overall interaction energy and electrostatic part of the interaction energy for the S-H bond in B2H6…B3S3X3 complexes are presented in Table 6.
Decomposition of two-bodied interaction energies within the IQA framework for the B3S3X3…B2H6 (X = H, F, Cl, CN, Me) systems at M062X/6-311G(d,p) level of theory (in a.u).
Positive
Positive
Exchange-interaction ratio (EIR)
The exchange-interaction ratio (EIR) is expressed as: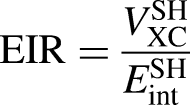
Because
For 0 < EIR ≪ 1 (when
Computed EIR values of S…H bonds in the B3S3X3…B2H6 systems indicate degree of ionic character.
Conclusion
Quantum chemistry exploration of replacement effects of hydrogens of borthiin with X = F, Cl, Me, CN on the interaction with B2H6 at M062X/6-311G(d,p) level of theory exposed more significance interaction energetically, in the presence of methyl group. largest electronic spatial extent value was found in the presence of Cl substituent. The most polarity of these complexes was belonged to CN group. Molecular orbital analysis revealed decreasing of HOMO-LUMO gap values as: X = F > H > Me > Cl > CN. The MEP maps obviously exemplified negative areas S atom of B3S3H3 and a positive area around bridged hydrogen of B2H6 molecule. The CDA outcomes expressed larger electrons donated from B3S3X3 fragment to the diborane molecule in the presence of X = H, F, Me. In contrast, greater electrons donated from diborane to B3S3X3 fragment in presence of X = Cl, CN. Computed EIR values of S…H bonds in the B3S3X3…B2H6 systems indicated degree of ionic character.
Footnotes
Ethical approval
Not applicable.
Consent to participate
All the co-authors consent to participate.
Consent to publish
All the co-authors consent to publish
Author's contribution
Reza Ghiasi: Supervision, Conceptualization, Data curation, Investigation, Formal analysis
Nahid Shajari: Methodology, Data curation, Writing, Review and Editing
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
Data will be made available on request.