
Abstract
Escherichia is one of the most studied bacterial genera and includes several clinically significant pathogenic species. Unique strategies have evolved in members of this genus to allow their survival within different niches. Although initially the Escherichia cryptic clades (CCs) I–VIII were identified as environmental bacteria, they have been found as commensals and opportunistic pathogens in animals and humans. Notably, members of the Escherichia CCs are commonly mistaken for E. coli when using standard microbiologic techniques for species identification. We used whole-genome sequencing to correctly identify E. marmotae—a member of the Escherichia CCs—that was initially misidentified as E. coli as the causative agent of a urinary tract infection (UTI) in a female dog. The isolate had phenotypic and genotypic resistance to cefalexin—an antibiotic used commonly for the treatment of uncomplicated UTIs—and carried a set of virulence factors also present in uropathogenic E. coli, which would explain the capacity of the organism to colonize the urinary tract. As a relatively new species that is phenotypically very similar to E. coli, it is quite possible that E. marmotae infections may be underreported or misidentified. We hope that our work alerts veterinarians to consider E. marmotae as a possible cause of UTIs in dogs, especially because this could affect treatment decisions. Additionally, our findings broaden the current understanding of the E. marmotae ecology and host range.
The genus Escherichia is known for the type species, E. coli, a very complex organism and one of the best studied among the family Enterobacteriaceae.13,57,69 The Escherichia genus also contains lesser-known pathogenic species, including E. albertii,
28
E. fergusonii,19,24 and the Escherichia cryptic clades (CCs) I–VIII.
40
Cryptic Escherichia species, including E. marmotae, E. ruysiae, and E. whittamii are commonly isolated from various environmental sources,8,31,75,76 and can carry antimicrobial resistance (
E. marmotae was first identified incidentally in Himalayan marmots during a 2015 study of the prevalence of Yersinia pestis in wild marmots (Marmota himalayana) living along the Qinghai-Tibet Plateau, China.31,32 Since then, the organism has been found colonizing and acting as an opportunistic pathogen in humans, 62 and in a range of non-human hosts, such as small rodents, 85 cattle, 43 and avian species.55,64 E. marmotae has also been isolated from waterways and other natural environments.8,34
Here, we present the complete genomic characterization of an E. marmotae isolate (UGA-EM1) found to have caused a recurring clinical urinary tract infection (UTI) in a dog.
Materials and methods
Bacterial isolation and species identification
A clinical urine sample was collected by a private veterinarian in the state of Georgia, USA, and submitted to the Athens Veterinary Diagnostic Laboratory (AVDL) of the University of Georgia (UGA; Athens, GA, USA) for culture and antimicrobial susceptibility (
Antimicrobial susceptibility testing
The minimum inhibitory concentrations (MICs) of 19 antimicrobials were determined using the GH97 card in a Vitek2 instrument based on Clinical and Laboratory Standards Institute (CLSI) guidelines.16,17 Our testing included amikacin, amoxicillin/clavulanic acid, ampicillin, cefalexin, cefovecin, cefpodoxime, ceftazidime, ceftiofur, chloramphenicol, ciprofloxacin, doxycycline, enrofloxacin, gentamicin, imipenem, marbofloxacin, nitrofurantoin, polymyxin B, and trimethoprim/sulfamethoxazole.
DNA extraction and genomic library preparation
DNA was extracted (DNeasy UltraClean microbial kit; Qiagen), following the manufacturer’s instructions, from an overnight culture on prepared blood agar plates and incubated overnight at 37 ± 2°C. Short-read barcoded sequencing libraries were prepared (Nextera DNA flex library prep kit; Illumina) following the manufacturer’s specifications. Sequencing was performed (MiSeq sequencing platform and 2 × 150 bp chemistry; Illumina).
Bioinformatic analyses
The GalaxyTrakr bioinformatic platform
25
was used for raw-read processing and de novo assembly. First, paired-end raw reads were qualitatively checked using FastQC
Additionally, within the Galaxy platform, the de novo assemblies were fed to ABRicate (v.1.0.1)
61
to identify AMR and virulence genes using default settings, and with StarAMR
7
(v.0.11.0), a software tool that incorporates ResFinder,
83
to identify AMR genes. PointFinder
84
was used to detect chromosomal point mutations associated with AMR (PointFinder package
84
) and extrachromosomal replicons (PlasmidFinder
12
). Genomic islands were predicted using IslandViewer4.
6
Isolate sequences were deposited in GenBank (
Results
Clinical manifestations and preliminary pathogen identification
An 8-y-old, spayed female dog was taken to a private veterinary hospital in the state of Georgia (USA) in March 2021, with UTI signs (frequent urination, discomfort urinating, bloody urine, accidents in the house, and/or licking genitals). A urinary sample from this patient was submitted to the AVDL for bacterial culture and AMS testing. Traditional microbiologic culturing techniques isolated a bacterium that was identified as E. coli by MALDI-TOF MS. MIC testing performed according to CLSI16,17 detected resistance to cefalexin (
WGS confirms UGA-EM1 as E. marmotae
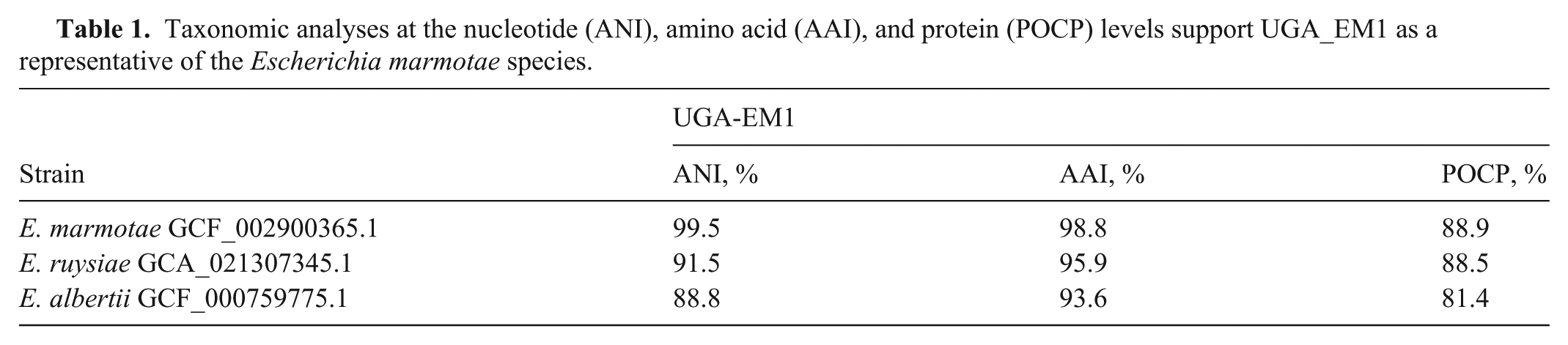
The short-read WGS of isolate UGA-EM1 yielded 1,147,199 raw reads, with 80% (917,370 reads) passing quality filtering (Phred score >30). The de novo assembly of the quality-filtered reads resulted in 408 contigs (≥1,000 bp), with an N50 value of 166,343 bp, and the longest contig comprising 623,175 bp. Several in silico taxonomic approaches contradicted the MALDI-TOF MS identification and classified UGA-EM1 as E. marmotae. First, a core-genome phylogenetic tree ( Fig. 1 ) including 14 NCBI reference genomes of the 10 known Escherichia species (E. albertii, E. coli [2 isolates]), E. fergusonii, E. marmotae, E. ruysiae, Escherichia sp001660175, Escherichia sp002965065, Escherichia sp004211955, and Escherichia sp005843885) proposed by the Genome Taxonomy Database, with Shigella dysenteriae as an outgroup, showed that UGA-EM1 appeared in the same clade as E. marmotae (Fig. 1). Similarly, an ANI analysis revealed that UGA-EM1 shared the highest genetic similarity (99.5% identity at nucleotide level) with E. marmotae YF8 (NCBI RefSeq GCF_029962465.1). In the AAI analysis of 4,175 coding DNA sequences conserved across the 10 Escherichia species, UGA-EM1 shared the highest amino acid similarity percentages with E. marmotae, followed by E. albertii and E. ruysiae ( Table 1 ). Finally, a POCP analysis performed on the genomes of UGA-EM1 and the 3 most closely related species according to core genome phylogeny (E. marmotae, E. ruysiae, E. albertii) also supported the identity of UGA-EM1 as E. marmotae (Table 1).
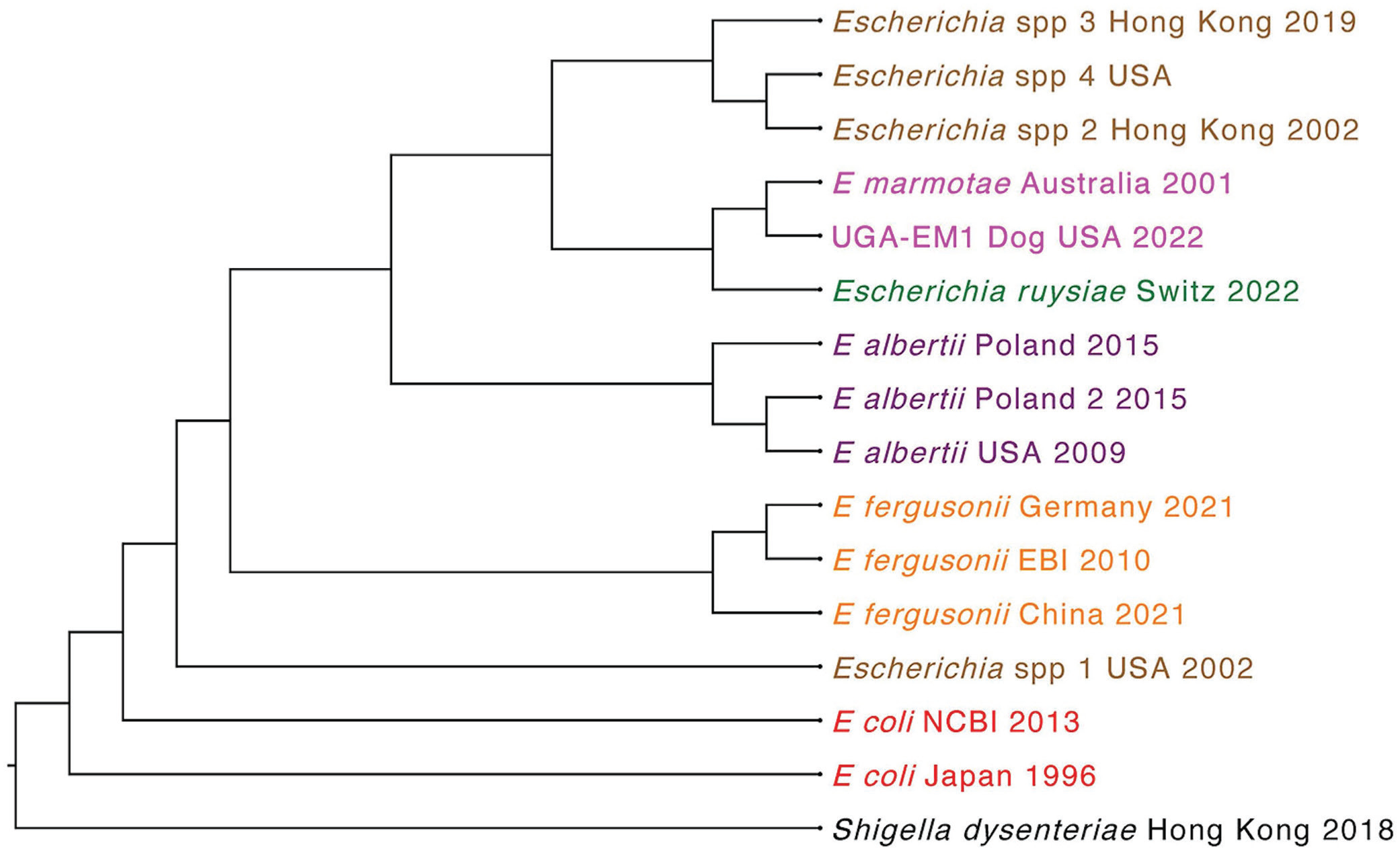
Escherichia phylogeny based on the core genomes of 14 representative isolates of the known Escherichia species: E. fergusonii (n = 3) in orange, E. coli (n = 2) in red, E. albertii (n = 3) in purple, E. ruysiae (n = 1) in green, Escherichia spp. (n = 4) in brown, and E. marmotae in pink (n = 1). Shigella dysenteriae reference genome was used as outgroup. UGA-EM1, in pink, clustered with E. marmotae. The tree was constructed using the maximum-likelihood algorithm in Parsnp (v.1.1) and visualized in Geneious Prime (v.2025.0).
Taxonomic analyses at the nucleotide (ANI), amino acid (AAI), and protein (POCP) levels support UGA_EM1 as a representative of the Escherichia marmotae species.
Genomic characterization of UGA-EM1
The UGA-EM1 genome was estimated at 4,620,417 bp and was assembled into 408 contigs. The length cutoff for the longest contigs that contain 50% of the total genome length or N50 was 166,346 bp. The genome has a GC content of 50.4%. No plasmids were detected in this isolate. The presence of other mobilizable elements was investigated and resulted in the deciphering of 41 genomic islands mostly associated with virulence genes (
Fig. 2
). UGA-EM1 carries a set of 57 virulence genes (
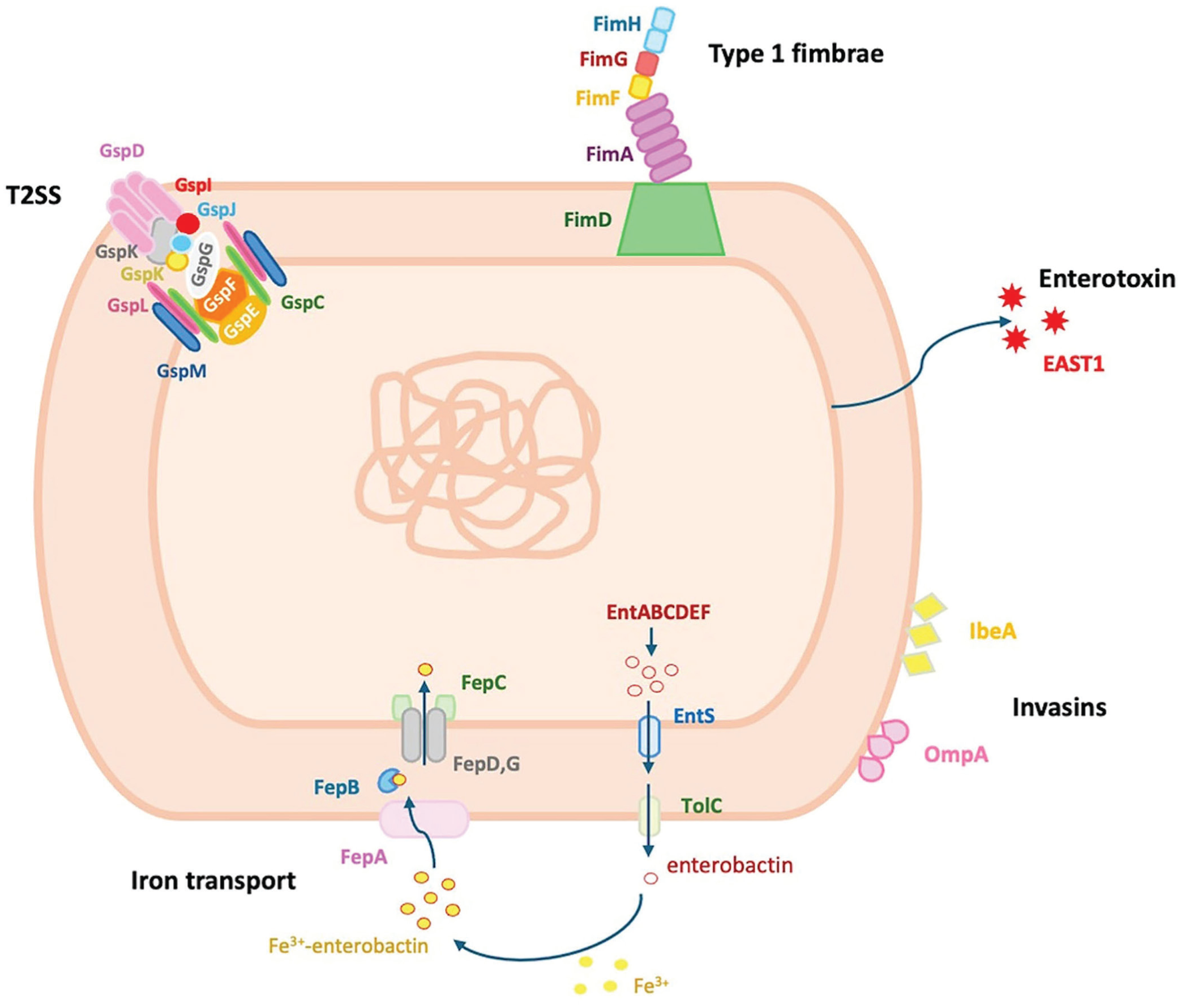
Schematic representation of the functional virulence mechanisms in Escherichia marmotae strain UGA-EM1: T2SS (gsp operon), type 1 fimbriae (fim operon), enterotoxin EAST1, invasins IbeA and OmpA, and 2-step enterobactin-mediated transport (fep and ent operons).
In the AMR genotype of UGA-EM1, we found a single blaEC gene, coding for beta-lactamase resistance. We also detected point mutations in parE (I355T) and parC (S57T) associated with fluoroquinolone resistance, and uhpT (E350Q) associated with fosfomycin resistance. MIC results showed that UGA-EM1 was susceptible to fluoroquinolones (enrofloxacin, marbofloxacin); hence, this point mutation was not sufficient to confer phenotypic resistance when analyzed using CLSI MIC interpretations. 16
Our phylogenetic analysis compared UGA-EM1 and 112 other E. marmotae complete genomes of isolates recovered from mammals, birds, humans, and the environment originating from different years and geographic regions. This broader comparative analysis identified 9 distinct genomic clusters (
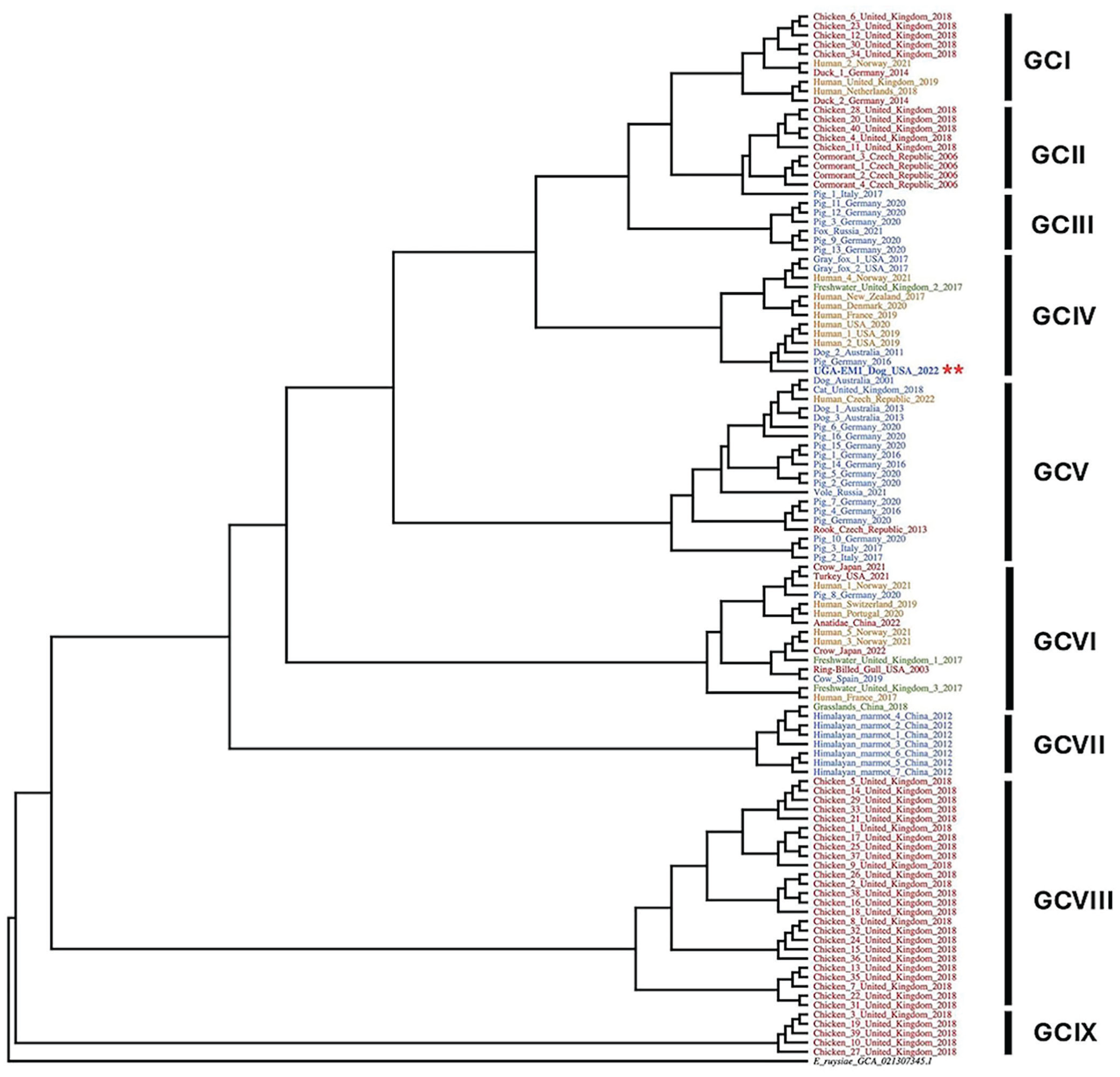
Escherichia marmotae phylogeny based on the core genomes of 112 representative E. marmotae strains and E. ruysiae (NCBI GCA21307345) as outgroup (black). Strains are color-coded by host, with 39 mammal isolates in blue, 52 avian isolates in red, 17 human isolates in orange, and 4 environmental (3 freshwater and 1 grassland) isolates in green. UGA-EM1 is in bold blue and carries red asterisks. Nine distinct genetic clusters (GCs) are identified (GCs I–IX). The tree was constructed using the maximum-likelihood algorithm in Parsnp (v.1.1) and visualized in Geneious Prime (v.2025.0).
Discussion
UTIs are common in female dogs, such as the subject of our study, given the nature of their anatomy; a short urethra and the proximity of the rectum provides an easy pathway and a large source of bacteria to the bladder. 45 A simple, uncomplicated UTI can occur sporadically in an otherwise healthy animal. 78 The presence of comorbidities, such as diabetes mellitus, urinary or reproductive tract infections, conformational abnormalities, and/or 3 or more episodes per year indicates a complicated case. 78 E. coli is the most common bacterial pathogen causing urinary tract infections in both uncomplicated and complicated cases. 68 Although E. marmotae are typically found in the natural environment, 75 they have also been reported to cause sepsis, spondylodiscitis, and UTIs in humans. 62 E. marmotae were also isolated from the ears of 2 dogs (NCBI GCA_021373445.1, NCBI GCA_021373315.1) and in the expressed milk from a dog in Australia (NCBI GCA_021373435.1). To determine the incidence of E. marmotae in canine populations, we conducted a systematic literature search across multiple high-impact scholarly databases, including PubMed/MEDLINE, CAB Abstracts, Web of Science, and Google Scholar. Utilizing the UGA institutional access to comprehensive biological and veterinary repositories, we did not identify any cases of canine UTIs attributable to E. marmotae. E. marmotae can be misidentified as E. coli in standard diagnostic workflows because of overlapping phenotypic profiles 62 ; therefore, its true clinical prevalence remains uncharacterized and its prevalence in UTIs is unknown. 75
MALDI-TOF MS is used in both veterinary and human clinical settings for the rapid identification of bacteria, supporting earlier treatment decisions and the timely implementation of outbreak mitigation strategies.47,62 Although the use of MALDI-TOF MS has been very beneficial in obtaining a microbiologic identification within 24 h, 11 certain limitations can hinder accurate identification, such as the absence of some species in the database or the inherent similarity between closely related species.14,18,54 In 2021, the first E. marmotae spectrum was added to the Bruker MALDI-TOF MS database (v.L2020 9607MSP); however, that spectrum was unable to reliably distinguish E. marmotae from E. coli. 62 A 2025 publication has reported the identification of E. marmotae with reliable identity scores (above the 2.0 threshold) using additional spectra that align more closely with typical E. marmotae profiles (7,260–7,268 m/z) using the Bruker system. 44 The initial identification of isolate UGA-EM1 was performed in 2021, using bioMérieux Vitek MS, knowledge base v.3.2, and did not include E. marmotae, which likely explains its misidentification as E. coli.
Our case is a clear example of E. marmotae preliminary misclassification, which was only corrected via WGS. Misclassification likely contributes to the fact that little is known about the epidemiology of E. marmotae in the environment or its role in a mammalian host (as either a commensal or an opportunistic pathogen), making this bacterium a potential public health concern.
The UGA-EM1 genome has a size comparable to other E. marmotae and does not harbor any plasmids, which is uncommon for this species.9,31 Nevertheless, UGA-EM1 carries a set of virulence genes in its chromosome that would facilitate colonization of the host urinary tract. Most of the virulence genes in UGA-EM1 are organized in operons such as the fae and the fim operons involved in the development of 2 different types of fimbriae. The fae operon encodes components of F4 fimbriae in enterotoxigenic E. coli strains. 82 These fimbriae are important adhesive structures that allow the bacteria to attach to the intestinal epithelium of the host. 15 A complete faeABCDEFGHI operon generally contains 8–9 genes. 1 Given that UGA-EM1 appears to be missing faeA (a major structural subunit of the F4 fimbrial shaft 74 ) and faeG (the adhesin that binds to host epithelial receptors 80 ) indicates that the isolate will not be able to assemble functional F4 fimbriae. However, UGA-EM1 carries a complete fimABCDEFGHI operon that encodes the type 1 fimbriae (or pili), a distinct set of adhesive surface structures different from F4 fimbriae. 15 Type 1 fimbriae in Escherichia species play a significant role in adhesion to host cells, particularly in the urinary tract and intestinal epithelium. 65
Additionally, the UGA-EM1 genome harbors the csgDEFG operon, part of the curli biogenesis system in several Enterobacteriaceae species. 3 Curli are extracellular amyloid fibers that contribute to biofilm formation, adhesion to surfaces, and host colonization. 3 In uropathogenic E. coli, the presence of csg genes has been associated with biofilm formation and promoted pathogenicity. 63 The curli system is encoded by 2 operons: csgBAC, which encodes the structural curli components, and csgDEFG, which encodes the regulatory and secretion machinery. 46 The fact that UGA-EM1 lacks the csgBAC operon suggests that curli subunits will not be synthesized even when their regulatory and secretion systems are present, as has been reported in Salmonella. 46
UGA-EM1 also contains the chu, ent, and fep operons, all involved in iron acquisition under iron-limiting conditions.2,63 The urinary tract is a particularly nutrient-poor environment, including limited iron availability. 35 Although the ent and fep operons are widely conserved across many Escherichia species,53,60 the chu operon is less broadly distributed but is still present in several gram-negative beta-proteobacteria, particularly those with pathogenic potential.20,56 These 3 operons have been described in E. marmotae. 85 These sets of genes would allow iron acquisition by UGA-EM1 and help establish a successful infection, as has been suggested for uropathogenic E. coli. 79 The chu operon, typically organized as chuASTUVWXY, 70 enables the use of heme from the mammalian host and can be found in adherent-invasive E. coli and enterohemorrhagic E. coli.38,63 The UGA-EM1 chu operon appears to be incomplete given that it lacks genes chuA and chuY. It has been suggested that chuY has an accessory or regulatory role38; therefore, deletion of this gene does not abolish the system’s function. 29 The chuA gene encodes a TonB-dependent outer membrane receptor that mediates heme transport from the environment into the periplasm. 20 Without chuA, the downstream components of the chu operon remain functional but do not have access to heme substrates 20 necessary to efficiently utilize heme as an iron source.
The ent and fep operons work together as a 2-part iron acquisition system based on the siderophore enterobactin.30,41 The ent genes encode enzymes involved in the biosynthesis of enterobactin, whereas the fep genes encode proteins responsible for the uptake and transport of iron-bound enterobactin into the bacteria. 30 UGA-EM1 most likely relies on this system for iron uptake.
UGA-EM1 also carries the gspCDEFGHIJKLM operon, conserved in many Escherichia species (in particular pathogenic species), which encodes the core components of the T2SS, and is required for the secretion of heat-labile enterotoxin—an important virulence determinant of enterotoxigenic E. coli.67,81 This system is also responsible for the secretion of folded proteins, such as enzymes, and other factors important for virulence, environmental survival, and nutrient acquisition.67,81 The only toxin we identified in the genome of UGA-EM1 was the enteroaggregative E. coli heat-stable enterotoxin (EAST1). 36 However, EAST1 is a very small peptide toxin that does not require periplasmic folding and transportation through T2SS. 22 EAST1 is produced by a subset of enteroaggregative E. coli strains and has been found in several E. coli pathotypes, including diffusely adherent E. coli, enterotoxigenic E. coli, and enteropathogenic E. coli.22,37,48 Although EAST1 is typically absent in non–E. coli species, the astA gene, which encodes the production of EAST1, 36 was found in the genomes of all the E. marmotae isolates included in our study, indicating that EAST1 is an important and conserved virulence gene in E. marmotae.
UGA-EM1 also carries 2 surface-exposed adhesin/invasion proteins encoded by the ibeA and ompA genes. In E. coli, both IbeA and OmpA mediate direct binding to specific host receptors, facilitating bacterial colonization of extraintestinal sites specifically. 48 IbeA is critical in helping neonatal meningitis-causing E. coli to cross the blood–brain barrier during neonatal brain infection, 57 whereas in avian pathogenic E. coli, IbeA contributes to the ability to cause septicemia and CNS infection in poultry. 26 OmpA is a multifunctional protein that promotes structural stability while facilitating adhesion, immune evasion, barrier penetration, and the impairing of immune cells, making it essential for full virulence in many E. coli pathotypes.42,50,66,72
UGA-EM1 had phenotypic and genotypic resistance to cefalexin, a beta-lactamase first-generation cephalosporin used to treat a variety of bacterial infections. 5 This group of beta-lactamases is distributed among most bacterial members of the order Enterobacterales, is primarily chromosomally encoded, and has a narrower hydrolytic spectrum than found encoded in plasmids. 58 Cefalexin is generally considered the first-line antibiotic for UTIs in dogs. 33 Therefore, it was the likely treatment of choice for the patient in our case, and that the recurrence of the UTI was the result of the resistance of UGA-EM1 to this antibiotic.
Finally, we examined the genetic relatedness of UGA-EM1 with other E. marmotae isolates. Overall, the phylogenetic analysis grouped the strains into 9 GCs, which were not strictly associated with host, geographic region, or year of collection, as observed previously. 9 This finding suggests that E. marmotae is not constrained by host-specific barriers and is capable of thriving across diverse ecologic environments. UGA-EM1 clustered with 3 other E. marmotae isolated from different hosts at very distant geographic locations, evidence that UGA-EM1 belongs to a lineage that is both geographically widespread and host-flexible. Global travel facilitates the spread of bacteria, such as UGA-EM1, by enabling the rapid movement of people, animals, goods, and their microbiota across continents. Travelers may unknowingly carry and disseminate both commensal and pathogenic bacteria, including strains harboring AMR genes.21,77 International travelers often acquire resistant bacteria during their trips, especially through food, water, or healthcare exposure, and may bring them back to their home countries, contributing to the global spread of resistant pathogens. 23 Among the animal isolates most genetically similar to UGA-EM1, one was collected from expressed milk fluid in an Australian dog with a mammary gland infection in 2011 (NCBI GCA_021373435.1) and another from the feces of a wild boar in Germany in 2016. Other close matches were 3 human isolates in the United States, 2 from Pennsylvania in 2019 and 2020 (DBFCQN000000000, DBFDLG000000000), and 1 obtained in Missouri in 2020, but without information specifying the disease status of the host or the source of isolation (NCBI GCA_011751205.1). Finding closely related isolates in dogs, pigs, and humans points toward zoonotic potential or a shared source, as has been suggested for Enterobacteriaceae.52,59
Despite increasing recognition of E. marmotae as a pathogenic species, a significant gap remains in our understanding. Limited surveillance data and the infrequent identification of E. marmotae in clinical and environmental settings hinder efforts to determine its true prevalence, transmission dynamics, and reservoirs. This lack of insight is further complicated by the genetic similarity to E. coli, which may lead to underreporting or misidentification. Hence, a One Health approach and further research are needed to characterize its distribution, host range, and potential role in zoonotic transmission.
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387261461260 – Supplemental material for Characterization of Escherichia marmotae, a novel pathogen causing urinary tract infection in a dog
Supplemental material, sj-pdf-1-vdi-10.1177_10406387261461260 for Characterization of Escherichia marmotae, a novel pathogen causing urinary tract infection in a dog by Wendy Cuevas-Espelid, Sonsiray Álvarez-Narváez and Susan Sánchez in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank Nikki Shariat and Erin Lipp for their advice and guidance, and the UGA AVDL for technical assistance during the conduct of our work.
Data availability
The genome assembly of Escherichia marmotae is available in the GenBank repository BioProject PRJNA1150114.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.