
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome caused by genetic defects in cytotoxic lymphocyte function. Current therapies can control disease activity, but transplantation of allogeneic hematopoietic stem and progenitor cells (HSPCs) remains the only curative option and is associated with substantial risks. These limitations have accelerated development of genome editing approaches enabling precise correction of disease-causing mutations in autologous cells. Familial HLH (FHL) represents a compelling target for genome editing, but successful and safe clinical translation has remained challenging. Preclinical studies demonstrate that targeted editing of key genes, such as PRF1 and UNC13D, can restore cytotoxic function in HSPCs and T cells. Translation to the clinic, however, depends on multiple factors, including the choice of target cell population, the level of functional correction required, and gene-specific constraints such as locus complexity and regulation of gene expression. In this review, we synthesize current progress in genome editing for FHL and highlight critical biological and technical barriers to clinical implementation. We propose a conceptual framework for designing genome editing strategies tailored to FHL, emphasizing the alignment of editing platform, gene architecture, and cellular context to enable effective and clinically translatable therapies.
Keywords
INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome characterized by uncontrolled activation of cytotoxic T lymphocytes (CTLs), natural killer (NK) cells, and macrophages. Clinically, patients typically present with the triad of fever, splenomegaly, and cytopenia. 1 HLH is broadly classified into primary (inherited) and secondary (acquired) forms. Primary HLH usually results from germline defects that impair the cytotoxic function of NK cells and CTLs, whereas secondary HLH arises in the context of strong immune triggers such as infection, malignancy, or autoimmunity. Despite these distinct initiating events, both forms converge on a common immunopathological pathway marked by impaired cytotoxic lymphocyte function, persistent immune activation, and systemic cytokine-driven inflammation.2,3
Primary HLH encompasses a heterogeneous group of inherited immune dysregulation disorders, including familial HLH types 2 to 5 (FHL2-5), which classically refers to autosomal recessive defects affecting lymphocyte cytotoxicity. The historical designation FHL1 predates gene identification and is no longer used. These autosomal recessive disorders disrupt either perforin production (FHL2) or the exocytosis of perforin-containing cytotoxic granules (FHL3-5). The causative genes for FHL2-5 are PRF1, UNC13D, STX11, and STXBP2, respectively.2–4 Other genetic defects leading to the HLH phenotype include mutations in genes involved in lysosome-related organelle biogenesis and trafficking, including RAB27A, LYST, AP3B1, and AP3D1, or in immune regulatory genes such as SH2D1A, XIAP, NLRC4, CDC42, NBAS, and RHOG.2–8 In this review, we mainly focus on two classical FHL-associated disorders, FHL2 and FHL3.
The aforementioned genetic defects impair the ability of NK cells and CTLs to eliminate antigen-presenting target cells and thereby limit termination of immune activation. Under physiological conditions, cytotoxic lymphocytes form an immunological synapse with their target cells, followed by granule polarization, trafficking along microtubules, docking, priming, membrane fusion, and exocytosis of perforin- and granzyme-containing granules. Perforin then polymerizes and forms pores in the target membrane, enabling granzymes to enter the target cell cytoplasm and trigger apoptosis, thereby resolving the immune response. In HLH, however, target cells are not efficiently eliminated, antigen presentation persists, and CD8+ T cells continue to produce proinflammatory cytokines, particularly interferon-γ (IFNγ). Activated macrophages further amplify this response through secretion of IL-1β, IL-6, IL-18, and TNFα, resulting in the cytokine storm that underlies the clinical manifestations of HLH, including fever, cytopenia, hepatosplenomegaly, and multiorgan failure.3,4,9–17
At present, transplantation of allogeneic HSPCs remains the only curative treatment for primary HLH. Although outcomes have improved, with a reported 3-year overall survival of 77%, transplantation of HSPCs remains limited by donor availability, graft-versus-host disease, the patient’s condition at the time of transplantation, and the toxicity of conditioning regimens. 18 These limitations have driven interest in gene-based approaches aimed at correcting the underlying genetic defect in autologous cells while reducing treatment-associated risks.
ADVANCES IN GENE-BASED THERAPIES FOR FHL
While FHL is genetically heterogeneous, most gene-based therapeutic strategies have focused on PRF1 and UNC13D (Table 1). This focus reflects several factors, including the relatively high frequency of mutations in these genes, the availability of robust preclinical models, such as the Prf1−/− (perforin knockout, PKO) model for FHL2 and the Jinx mouse model for FHL3, and well-established functional assays to assess cytotoxicity, including degranulation and target-cell killing assays.19,21–23,27
Evolution of gene-based treatment strategies for FHL
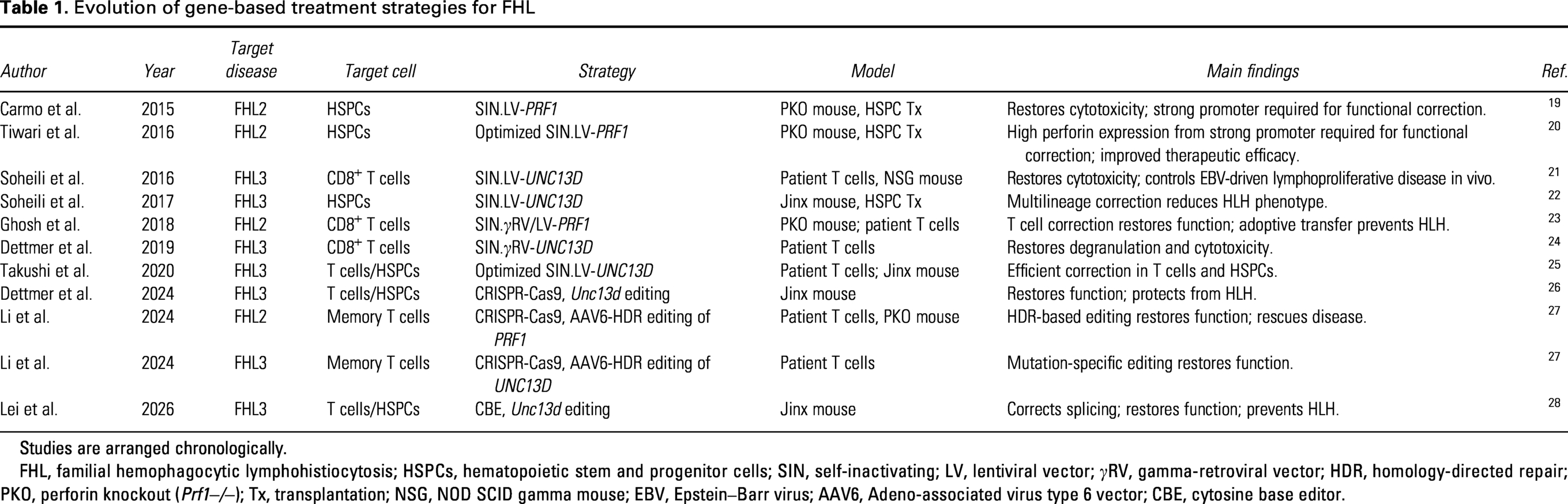
Studies are arranged chronologically.
FHL, familial hemophagocytic lymphohistiocytosis; HSPCs, hematopoietic stem and progenitor cells; SIN, self-inactivating; LV, lentiviral vector; γRV, gamma-retroviral vector; HDR, homology-directed repair; PKO, perforin knockout (Prf1−/−); Tx, transplantation; NSG, NOD SCID gamma mouse; EBV, Epstein–Barr virus; AAV6, Adeno-associated virus type 6 vector; CBE, cytosine base editor.
Early studies addressed the fundamental question whether restoration of cytotoxic lymphocyte function is sufficient to reverse FHL pathology. In FHL2, lentiviral-mediated addition of the human PRF1 gene to murine PKO CD8+ T cells restored cytotoxicity and prevented FHL development following adoptive transfer. 23 Similarly, PRF1 addition to HSPCs of PKO mice restored perforin expression and ameliorated disease manifestations in this preclinical model. 19 These findings established that correction of CD8+ T cells function alone can be sufficient to control disease, although durability likely depends on the targeted cell compartment.
Parallel work in preclinical FHL3 disease models demonstrated that retroviral or lentiviral addition of human UNC13D restores cytotoxic function in both Jinx mouse- and patient-derived T cells.21,22,24,25 Transplantation of lentiviral vector transduced HSPCs resulted in multilineage reconstitution and attenuation of HLH manifestations. 22 Collectively, these studies established two key principles: first, that FHL is biologically reversible or preventable upon restoration of cytotoxic function, and second, that both mature lymphocytes and HSPCs are viable therapeutic targets.
Importantly, this therapeutic paradigm extends beyond classical FHL. Gene addition strategies have also been applied to syndromic HLH, such as XIAP deficiency, where lentiviral gene transfer restored immune function and normalized hyperinflammatory responses in preclinical models.29,30
Building on these gene addition strategies,31–34 the field has increasingly shifted toward genome editing approaches that enable precise correction at the endogenous locus. CRISPR/Cas9-based editing has been used to repair disease-causing mutations in both FHL2 and FHL3, restoring cytotoxic function in primary human T cells or, upon transplantation of gene-edited murine HSPCs, respectively.26,27 Notably, targeted repair of Prf1 in PKO memory T cells using homology-directed repair (HDR) restored cytotoxicity and prevented HLH-like disease in vivo. 27 Similarly, CRISPR-Cas9-mediated disruption of the disease-underlying Unc13d mutation in HSPCs and T cells restored immune function and protected against HLH in the preclinical Jinx model. 26 More recently, next-generation editing platforms have further expanded the therapeutic toolbox. In this context, base editing has been applied to disrupt a pathogenic cryptic splice site in Unc13d in Jinx mice, restoring correct splicing and cytotoxic function, as well as protecting these animals from disease. 28 These findings demonstrate the therapeutic potential of base editing approaches for the treatment of genetic hyperinflammatory disorders.
Despite these advances and the prospect of more precise and physiologically regulated therapies, the few genome editing approaches for FHL are still at a preclinical stage. In contrast, lentiviral gene addition remains the most clinically advanced platform in gene-based therapies for FHL, as illustrated by an ongoing phase I/II trial (NCT06736080) using autologous UNC13D-transduced CD34+ cells and T cells in FHL3 patients at the Necker Hospital in Paris.35,36
TARGET CELL POPULATION
A central unresolved question in genome editing strategies for FHL is which immune compartment must be corrected to achieve durable disease control. Because the primary defect lies in cytotoxic lymphocyte function, the main candidate targets are T cells, NK cells, and HSPCs.37,38
The strongest mechanistic evidence identifies impaired CD8+ T cells as the dominant drivers of established FHL pathology.38,39 This conclusion is supported by adoptive transfer studies, in which infusion of functional cytotoxic T cells reversed active HLH manifestations in both PKO and Jinx mouse models and provided protection against relapse.19,21,23,27,39–42 These findings highlight the central role of T cell cytotoxicity in controlling disease once hyperinflammation is established.
Within the T-cell compartment, long-lived memory T-cell subsets may represent an attractive target for genome editing approaches. In this context, autologous T-memory stem cells combine the self-renewal capacity of stem-like cells with the rapid effector potential of differentiated T cells, potentially enabling durable immune reconstitution following precise genetic correction. Recent studies demonstrated successful correction of PRF1 and UNC13D in autologous memory T cells, restoring cytotoxic function in vitro and in vivo. 27 These findings suggest that editing of long-lived T-cell populations may provide sustained therapeutic benefit while avoiding some of the challenges associated with HSPC transplantation and conditioning.
NK cells represent an additional, potentially complementary therapeutic target. Their role in early antiviral defense positions them upstream of CD8+ T-cell expansion, and impaired NK cell-mediated clearance may increase antigen burden and thereby amplify downstream CTL activation and cytokine production. In this context, NK cells may contribute not only to direct cytotoxic control of infected cells but also to early immune regulation by limiting persistent antigen presentation and excessive activation of adaptive immune responses. Despite this biological relevance, direct evidence demonstrating that NK cell-restricted correction is sufficient to prevent or reverse HLH remains lacking. One possible explanation is that most established HLH models are dominated by CTL-driven pathology once hyperinflammation is fully established, making therapeutic rescue by NK cells alone more difficult to demonstrate experimentally. In addition, durable and selective genetic modification of NK cells remains technically more challenging than editing of T cells or HSPCs, limiting the availability of dedicated NK cell–based preclinical studies. Consequently, while NK cells likely play an important upstream regulatory role in HLH pathogenesis, their precise therapeutic contribution in genome editing strategies remains incompletely defined and warrants further investigation.38,39
This uncertainty makes HSPCs an attractive target, as their modification enables multilineage correction and promises long-lasting effects.22,25,26 However, HSPC-targeted editing faces a particularly important clinical obstacle: despite improvements in newborn screening, the majority of pediatric FHL patients present with active, full-blown HLH at the time of diagnosis. The acute inflammatory state, together with the urgent need for aggressive immunosuppression, often makes HSPC mobilization and harvest difficult or infeasible. This underscores the need to develop strategies for readily accessible cell types, such as peripheral T cells. Platform optimization will therefore need to center on two parallel tracks: (i) refining editing tools in terms of delivery methods, editing efficiency, and safety for HSPC-based approaches applicable to patients who can be stabilized prior to harvest, and (ii) developing robust T cell-based correction protocols that can be deployed rapidly in an acute setting. Ex vivo editing of autologous T cells, particularly with non-viral delivery platforms, should therefore be prioritized given its potential compatibility with the clinical urgency of active HLH. 39
From a genome editing perspective, the choice of target cell population is constrained by differences in DNA repair biology. CRISPR-Cas9-mediated editing relies on endogenous DNA repair pathways following induction of DNA double-strand breaks (DSB). Non-homologous end joining (NHEJ) is highly efficient but can be erroneous, frequently resulting in insertion and deletion mutations (indels) at the target site. Accordingly, NHEJ-based strategies are typically used to disrupt coding or cis-regulatory sequences with high editing efficiencies, often exceeding 90% allele modification in both HSPCs and T cells. In contrast, precise sequence correction or targeted insertion of exogenous DNA generally requires HDR, which is largely restricted to cells in the S/G2 phase of the cell cycle. 43 Because long-term repopulating hematopoietic stem cells predominantly reside in a quiescent G0/G1 state, HDR efficiency in this compartment remains limited.44,45 In contrast, activated T cells are highly permissive to HDR, which has driven the development of HDR-based genome editing strategies targeting T cells.46,47
Accordingly, each target compartment presents distinct advantages and limitations. Genome editing of HSPCs offers the potential for a one-time, lifelong cure through multilineage reconstitution but requires conditioning. As demonstrated in clinical studies for hemoglobinopathies, disruption of cis-regulatory elements can be achieved at high, stable, and clinically well-tolerated levels.48–53 In contrast, HDR-based correction strategies in HSPCs remain challenging, and long-term safety considerations persist. Editing of mature T cells avoids myeloablative conditioning and may provide a more immediate and potentially safer intervention but does not correct defects in other cytotoxic compartments and may require repeated administration. Together, these findings support two complementary therapeutic strategies: effector cell correction, as highlighted by Booth et al., likely represents a bridge-to-transplant approach in critically ill patients, whereas multilineage correction via HSPC editing may provide long-term protection from hyperinflammation. 54 The aforementioned clinical study (NCT06736080), which incorporates both T cell- and HSPC-based strategies, further underscores that these approaches can be pursued in parallel or potentially combined.36,54
BIOLOGICAL THRESHOLDS FOR DISEASE CONTROL
A central question in the development of gene-based therapies for FHL is the level of corrected cells required to restore immune homeostasis. Evidence from preclinical and clinical studies indicates that therapeutic efficacy depends on achieving a threshold of functional immune reconstitution rather than complete hematopoietic replacement.19,55,56
In both PRF1 and UNC13D deficiency, studies in murine models have shown that relatively low levels of donor chimerism are sufficient to prevent HLH and restore immune control. Notably, approximately 20–30% graft-derived T cell chimerism was adequate to suppress disease manifestations, indicating that a limited fraction of functional cytotoxic lymphocytes can re-establish immune regulation.19,28,39,55 These findings support the concept of a functional threshold for cytotoxic activity, above which immune homeostasis can be maintained.
This principle has important implications for therapeutic design. Rather than aiming for complete correction of the hematopoietic system, genome editing strategies may prioritize achieving sufficient levels of functional correction to surpass this threshold. This is particularly relevant in the context of HDR-based genome editing, where editing efficiencies may be constrained by delivery, DNA repair pathways, or cell-intrinsic factors.
Clinical experience from other inborn errors of immunity further supports this concept. In adenosine deaminase–deficient severe combined immunodeficiency, Wiskott–Aldrich syndrome, and X-linked chronic granulomatous disease, partial chimerism of 5–20% has been sufficient to achieve durable immune reconstitution and meaningful clinical benefit.57–61 Hence, relatively low levels of corrected HSPCs can give rise to a sufficient pool of functional effector cells to restore immune competence.
Taken together, these findings establish a key design principle for genome editing in HLH: therapeutic success depends on reaching a threshold of approximately 20% of functional cytotoxic capacity rather than complete genetic correction. This threshold-dependent model provides a realistic and clinically relevant framework for evaluating genome editing strategies, particularly in settings where editing efficiency is inherently limited.
STRATEGIC CONSIDERATIONS FOR GENOME EDITING IN FHL
Genome editing strategies for FHL must be tailored to the biological and molecular characteristics of the target gene, most prominently PRF1 and UNC13D. These genes present distinct engineering challenges and therefore require different therapeutic design approaches (Fig. 1).
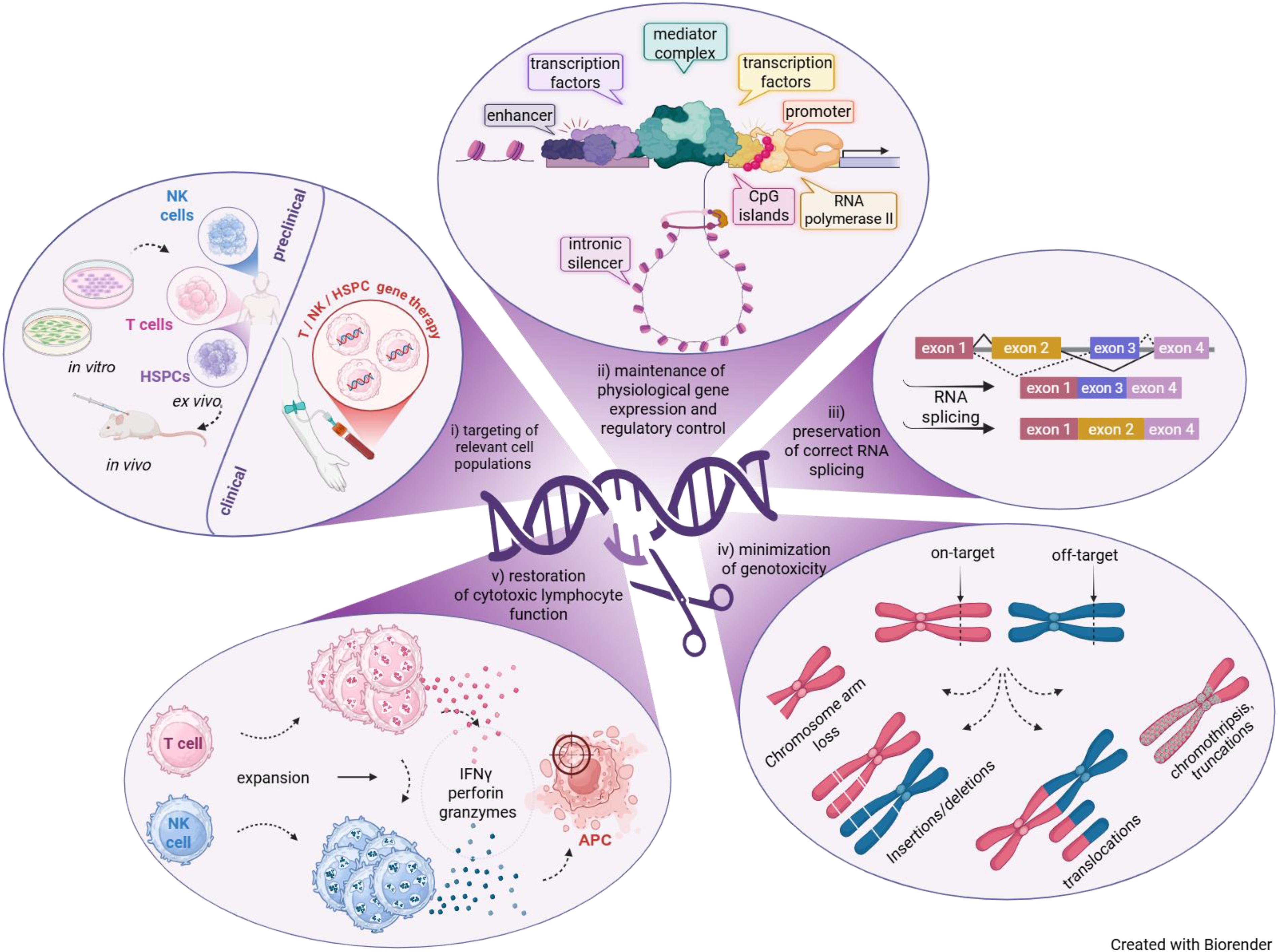
Design principles and biological requirements for genome editing strategies in FHL. Genome editing approaches for FHL must address multiple interconnected biological and technical design requirements. (i) Therapeutic strategies must target clinically relevant cell populations, including T cells, natural killer (NK) cells, and hematopoietic stem and progenitor cells (HSPCs), while enabling translation from preclinical validation toward clinical application. (ii) Genome editing strategies must preserve physiological gene regulation, including promoter activity, enhancer interactions, CpG island architecture, transcription factor binding, mediator complex recruitment, and intronic regulatory elements. This is particularly relevant for UNC13D, which is characterized by complex transcriptional regulation and frequent splice-altering mutations. (iii) Correct RNA processing and splicing must be maintained or restored to enable physiological gene expression. (iv) Genome editing strategies must minimize unintended on-target and off-target genotoxicity, including insertion/deletion mutations (indels), chromosomal translocations arising from concomitant cleavage of on-target and off-target sites, chromosome arm loss, and chromothripsis-related truncations. Chromothripsis refers to extensive chromosome fragmentation and aberrant reassembly following DNA damage. Red chromosomes harbor intended target locus, whereas blue chromosomes contain an off-target site. (v) Ultimately, successful genome editing should restore cytotoxic lymphocyte effector function, including perforin/granzyme-mediated target cell killing, thereby contributing to re-establishment of immune homeostasis and prevention of hyperinflammatory disease manifestations. Abbreviations: APCs, antigen-presenting cells; HSPCs, hematopoietic stem and progenitor cells; IFNγ, interferon-gamma; NK, natural killer.
At the genomic level, PRF1 is a relatively compact gene (∼6.4 kb) with 3 exons and limited structural complexity, whereas UNC13D represents a substantially larger and more complex locus (∼18 kb) with 32 exons, alternative regulatory elements, and a high prevalence of splice-altering mutations in FHL3 patients.62–67 In both genes, pathogenic variants are distributed across the locus.68–70 As a consequence, no single genome editing strategy is likely to be universally applicable across all HLH genotypes. These differences define distinct design constraints. For PRF1, the primary challenge lies in achieving sufficient levels of functional protein expression required for effective cytotoxic function, making expression control and locus context critical determinants of therapeutic success. 20 In contrast, for UNC13D, the dominant challenge is not expression level per se but the regulatory complexity of the gene, which complicates precise correction and faithful restoration of endogenous gene regulation.
Genome editing approaches broadly fall into two categories: mutation-specific repair and mutation-agnostic (universal) correction strategies. Mutation-specific repair aims to correct the endogenous gene at its native locus, thereby preserving physiological regulation. This approach is particularly relevant for splice-defective UNC13D variants, where restoration of correct RNA processing is essential.26,28 Similarly, targeted exon replacement strategies for PRF1 and UNC13D may enable correction of mutation hotspots while maintaining endogenous expression patterns. 27
In contrast, mutation-agnostic approaches aim to bypass the underlying mutation by introducing a functional gene copy. These include HDR-mediated targeted integration of a partial cDNA into an early exon of the endogenous locus, 27 targeted integration of an entire expression cassette into safe-harbor loci such as AAVS1 71 or targeted integration into an essential gene such as GAPDH, which enables efficient knock-in by coupling successful integration to cell survival.72,73 While essential gene-targeting strategies and safe harbor integration approaches can achieve high editing efficiencies, they do not recapitulate endogenous gene regulation. Moreover, a major limitation for precise HDR-based genome editing is the requirement for efficient delivery of donor templates. Increasing template size is associated with reduced HDR efficiency and increased cellular toxicity, particularly in primary human lymphocytes and HSPCs. 74 This constraint is especially relevant for UNC13D, where the large coding sequence and regulatory complexity challenge both viral and nonviral delivery platforms. While compact cDNA-based approaches may facilitate delivery, they may fail to fully restore endogenous transcriptional and post-transcriptional regulation.
Beyond target site selection, platform-specific constraints further shape therapeutic design. CRISPR-Cas9-mediated editing relies on the induction of DSBs, which can introduce genotoxic risks extending beyond small insertion/deletion mutations (indels). Increasing evidence demonstrates that nuclease-induced DNA repair can generate large deletions, chromosomal rearrangements, translocations, megabase-scale loss of heterozygosity, and, in rare cases, chromothripsis.75–80 Importantly, many of these structural alterations are not detected by standard short-range genotyping approaches and therefore represent a major safety concern for clinical translation. 81 Recent work further demonstrated that the extent and nature of genotoxicity strongly depend on both the editing platform and the targeted cell type. In this context, our laboratory systematically compared gene editing strategies for therapeutic correction of an FHL3-associated mutation and identified distinct platform- and cell type-specific genotoxicity profiles, including differences between nuclease-based editing and base editing approaches. 28 These findings emphasize that genome editing strategies cannot be evaluated solely based on editing efficiency but also require comprehensive assessment of genomic integrity in clinically relevant cell populations. These risks, together with the historical experience of insertional mutagenesis in retroviral gene therapy strategies, such as activation of the LMO2 proto-oncogene in X-linked severe combined immunodeficiency and clonal expansion events in Wiskott–Aldrich syndrome, have driven the field toward the development of more precise editing strategies that preserve genomic integrity.82,83
To mitigate the risks associated with DSB-based editing, alternative genome editing technologies are being actively developed. Base editing and prime editing enable precise sequence modification without requiring the introduction of DSBs and have shown promising results in preclinical models as well as early clinical translation.28,53,84–87
Base editing has emerged as a particularly attractive strategy for FHL. By enabling precise nucleotide conversion, base editing is well suited for correcting UNC13D splice-site mutations and a substantial fraction of single-nucleotide variants, both of which are highly prevalent in FHL3 patients. In the Jinx model, cytosine base editing has been successfully applied to disrupt a pathogenic cryptic splice site, restoring correct splicing, cytotoxic function, and protection from HLH in vivo. However, the occurrence of platform- and cell type-specific chromosomal rearrangements should not be overlooked. 28
Prime editing approaches, including twin and quadruple prime editing, further extend these capabilities to more complex sequence modifications. Recent developments have demonstrated targeted insertion of large DNA sequences of up to 26 kb, suggesting potential applicability to large genes such as UNC13D.88–90 However, the performance of these approaches in clinically relevant primary cells and their ability to fully restore physiological gene function remain to be established.
Taken together, these considerations highlight a central design principle for genome editing in HLH (Fig. 1). Therapeutic strategies must be tailored to the specific gene architecture, mutation type, and cellular context. For UNC13D, gene size, mutation diversity, and regulatory complexity impose significant constraints on current technologies. For PRF1, the smaller gene size expands the range of feasible editing strategies, but achieving sufficient expression levels remains critical. Ultimately, successful genome editing in FHL will require aligning the editing platform with both the molecular properties of the target gene and the functional requirements of cytotoxic lymphocytes.
CHALLENGES REMAINING FOR EFFICIENT TRANSLATION
Despite rapid progress, several key barriers continue to limit the clinical translation of genome editing strategies for FHL. These challenges span conditioning toxicity, cell-intrinsic limitations, and manufacturing complexity.
Toxicity of conditioning regimens
Pediatric patients represent the majority of individuals undergoing treatment for FHL. 91 This population is particularly vulnerable to the toxic effects of conventional conditioning regimens, which are associated with significant morbidity and mortality, including infertility and secondary malignancies.18,92 Although reduced-intensity conditioning has improved survival, toxicity remains a major limitation. 93 Alternative approaches, such as non-genotoxic conditioning using CD45- or CD117-targeted antibody–drug conjugates, have shown promising results in early studies and may enable safer engraftment of gene-edited HSPCs. 94 In parallel, genome editing–based strategies such as epitope editing may allow the generation of “stealth” HSPCs that are selectively enriched in vivo, potentially reducing the need for cytotoxic conditioning.95–97
Impact of inflammatory disease state on HSPC fitness and editing efficiency
A frequently overlooked challenge is the physiological state of the patient. In contrast to preclinical studies performed with healthy donor cells, HSPCs for autologous therapies are derived from patients with ongoing or recent hyperinflammation. Studies in related disorders, such as chronic granulomatous disease and sickle cell disease, have shown that inflammatory stress alters HSPC biology, including transcriptional programs associated with TNFα signaling, interferon responses, and cell cycle regulation.98,99 These changes can impair stem cell fitness, promote lineage skewing, and reduce engraftment potential despite successful genetic correction. Moreover, because genome editing relies on endogenous DNA repair pathways, inflammation-induced alterations in DNA damage responses may affect both editing efficiency and genomic stability. 100 An additional challenge relates to the delivery of genome editing reagents. Ex vivo genome editing of HSPCs still predominantly relies on electroporation-based delivery, which itself can impair cell fitness and viability. This issue may become particularly relevant in cells already exposed to inflammatory stress. Whether alternative delivery systems, such as lipid nanoparticle (LNP)-based approaches, can mitigate these limitations while maintaining efficient editing remains to be established. Together, these findings indicate that the inflammatory context of HLH patients represents a critical and underappreciated determinant of therapeutic outcome.
Manufacturing complexity and time constraints
Current genome editing approaches rely on complex ex vivo workflows, including mobilization and collection of patient cells, genetic modification, and reinfusion following conditioning. These processes are technically demanding, resource-intensive, and costly. 101 Importantly, HLH is characterized by rapid disease progression and narrow therapeutic windows, making delays associated with manufacturing particularly problematic. 102 This logistical complexity represents a major barrier to broad clinical implementation.
Emerging in vivo genome editing strategies
To overcome the limitations associated with ex vivo cell manufacturing, in vivo genome editing approaches are being actively explored. These strategies aim to directly modify HSPCs within their native niche, thereby eliminating the need for cell harvest, prolonged ex vivo manipulation, and reinfusion following conditioning. Proof-of-principle has already been demonstrated in recent clinical and preclinical studies, including LNP-mediated base editing in the liver of a neonate with CPS1 deficiency, showing clinical benefit and short-term safety. 84
Multiple delivery platforms are currently under investigation for in vivo targeting of hematopoietic cells, each presenting distinct advantages and limitations. LNP-based RNA delivery systems offer transient expression, low risk of genomic integration, and scalable manufacturing, making them particularly attractive for nonviral delivery of genome editing reagents. 103 In addition, LNP-mediated delivery has demonstrated efficient and minimally toxic editing of human HSPCs both ex vivo and increasingly also in vivo.104,105 However, efficient and selective targeting of long-term repopulating hematopoietic stem cells remains challenging.
Virus-like particles (VLPs) provide an alternative transient delivery strategy by enabling efficient transfer of genome editors while avoiding persistent vector expression. Their favorable transient expression kinetics may reduce prolonged nuclease exposure and thereby potentially limit genotoxicity.106–108 In contrast, adeno-associated viral vector (AAV)-based systems provide highly efficient nucleic acid delivery and have demonstrated promising HSPC targeting efficiencies in preclinical studies.109,110 However, concerns remain regarding payload limitations, prolonged expression of editing components, and potential immunogenicity. 111 While all of these in vivo approaches hold considerable promise for simplifying treatment and improving scalability, challenges related to delivery efficiency and imperfect target cell specificity continue to restrict their full therapeutic potential.
Importantly, the inflammatory environment characteristic of HLH may pose an additional barrier to successful in vivo genome editing. Active or recent hyperinflammation can alter HSPC fitness, cell cycle state, and DNA repair responses, potentially affecting editing efficiency, genomic integrity, and long-term engraftment of edited cells. 112 Moreover, inflammatory activation of innate immune pathways may influence vector uptake, persistence, and immune clearance of delivery systems. Consequently, strategies successful in healthy preclinical settings may not directly translate to patients with active inflammatory disease.
Together, these emerging technologies hold considerable promise for simplifying genome editing therapies and improving scalability. However, further optimization of delivery specificity, editing efficiency, immunogenicity, and performance under inflammatory conditions will be required before in vivo genome editing approaches can become clinically applicable for HLH.
CONCLUSION
Recent advances have established genome editing as a promising therapeutic strategy for primary HLH, particularly for disorders caused by PRF1 and UNC13D deficiency. Preclinical studies demonstrated that restoration of cytotoxic lymphocyte function can reverse or prevent hyperinflammatory disease manifestations, supporting the concept that HLH is biologically amenable to targeted genetic correction. Importantly, both HSPC- and T cell–based approaches have shown therapeutic potential, with each strategy offering distinct advantages depending on the target gene, disease stage, and clinical context.
At the same time, these studies have highlighted that successful genome editing in HLH is not a one-size-fits-all approach. Rather, therapeutic design must account for gene-specific constraints, including the regulatory complexity and splice sensitivity of UNC13D, as well as the stringent expression requirements of PRF1. In addition, the choice of target cell population critically influences editing feasibility, durability of correction, and translational applicability.
Future progress in the field will depend not only on improvements in editing efficiency and genomic safety but also on optimization of delivery systems, conditioning strategies, and target cell selection. In particular, advances in base editing, prime editing, and in vivo delivery technologies may help overcome current limitations associated with DSB-based editing and ex vivo cell manufacturing. Equally important will be a better understanding of how inflammatory disease states influence HSPC fitness, DNA repair responses, and long-term engraftment of edited cells.
Taken together, genome editing for primary HLH is transitioning from proof-of-concept toward early clinical translation. Continued integration of genome engineering technologies with disease-specific biological insights will be essential to develop safe, durable, and broadly accessible therapies for patients with HLH.
Footnotes
ACKNOWLEDGMENTS
The authors thank the members of our laboratory for fruitful discussions. The authors further acknowledge the use of AI tools (ChatGPT) for literature research and assistance in improving the readability of the article. All references, factual statements, and literature citations were independently verified by the authors.
AUTHOR DISCLOSURE STATEMENT
T.C. is an advisor to AaviGen, AstraZeneca, Novo Nordisk, and T-knife Therapeutics. All other authors declare the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
FUNDING INFORMATION
The authors are supported i.a. by the European Union (Horizon Europe,