
Abstract
Chimeric antigen receptor (CAR) T-cell therapy has transformed hematological cancer care, yet variability in efficacy, durability, and safety cannot be explained solely by antigen selection or patient factors. We propose that manufacturing platforms are active biological determinants of outcome. Viral vectors, used in all licensed products, provide stable genomic integration and durable expression but are limited by cost, cargo capacity, and centralized production. Nonviral strategies, including transposons, CRISPR knock-ins, and messenger RNA delivery, enable faster, less-expensive manufacturing with larger payloads, while introducing distinct safety and persistence profiles. This review presents a three-layer mechanistic framework that reframes manufacturing as biology: integration biology determines genomic risk and transgene stability; clonal fitness shapes persistence, dominance, and exhaustion; and epigenomic imprinting, influenced by gene transfer method, cytokines, and culture stress, preconfigures functional trajectories. Clinical observations link platform choice to immune recovery, where prolonged B-cell aplasia and delayed T-cell reconstitution contribute to infection-related nonrelapse mortality, and hematopoietic reserve at apheresis emerges as a practical predictor. Finally, manufacturing is positioned as the key to democratizing cell therapy. Decentralized, nonviral production aligned with regulatory standards may enable equitable access and transition CAR-T therapy from innovation to sustainable global care.
Keywords
KEY POINTS
Manufacturing is destiny: Platform choice imprints therapeutic fate through a three-layered mechanistic framework including integration biology, clonal fitness, and epigenomic imprinting that together govern persistence, exhaustion, and long-term safety. Viral vectors underpin all licensed products, delivering stable expression and durable activity but constrained by cost, cargo limits, centralized supply chains, and lifelong vigilance for insertional events. Nonviral platforms (transposons, CRISPR knock-ins, messenger RNA) expand payload capacity, reduce costs, and accelerate production, while introducing platform-specific risks (e.g., clonal dominance, editing-related genomic lesions, underpersistence) that require tailored surveillance. Clinical benchmarks from mature viral-vector trials define efficacy and toxicity standards; early nonviral signals are promising but heterogeneous, and cross-trial comparisons must remain heuristic given differences in design, endpoints, and follow-up. Immune recovery is platform-dependent: prolonged B-cell aplasia and delayed T-cell reconstitution drive infection risk now the leading cause of nonrelapse mortality; hematopoietic reserve at apheresis is an actionable predictor of delayed reconstitution. Democratization will hinge on nonviral and point-of-care manufacturing integrated with regulatory harmonization, enabling equitable access and consistent quality across diverse health care settings.
INTRODUCTION
Chimeric antigen receptor (CAR) T-cell therapy has redefined the treatment of hematological malignancies, delivering unprecedented remission rates in relapsed or refractory B-cell leukemias, lymphomas, and multiple myeloma.1–3 Despite these transformative successes, wide variability in efficacy, durability, and toxicity has become apparent across trials and real-world settings. 4 Increasingly, it is clear that these outcomes are not solely dictated by the CAR construct or disease biology, but by the manufacturing platforms and processes that generate the therapeutic cells.5–7
The historical trajectory of CAR-T development underscores this point. The first generation of licensed products, including tisagenlecleucel, axicabtagene ciloleucel, and brexucabtagene autoleucel, was all built on viral vector technologies, primarily γ-retroviruses and lentiviruses. 8 These vectors offered stable integration and predictable transgene expression and were supported by decades of preclinical and clinical experience. Their success cemented the viral paradigm as the field’s foundation. Yet, the same reliance has also exposed critical vulnerabilities: the high cost of GMP-grade viral vector production, supply-chain constraints, long turnaround times, and ongoing concerns over insertional mutagenesis.5,7 These bottlenecks have restricted patient access, particularly outside of high-income countries, and highlight how manufacturing choices can directly shape the global reach of cellular therapies.
Emerging nonviral systems, including transposons such as Sleeping Beauty and PiggyBac, CRISPR-mediated knock-ins, and messenger RNA (mRNA) platforms, are now challenging this status quo. By eliminating dependence on large-scale viral vector production, these approaches promise reduced costs, faster manufacturing, and in some cases safer genomic profiles.6,9,10 They also enable larger or more complex genetic payloads, including multigene cassettes and switchable synthetic circuits, that would be difficult to accommodate in viral systems. However, enthusiasm is tempered by fresh uncertainties. Recent reports of PiggyBac-associated CAR-T-derived lymphomas driven by clonal dominance, 11 variable persistence with mRNA platforms, 12 and off-target concerns with CRISPR 13 highlight the risks of moving too quickly without a mechanistic understanding of how these technologies reshape T-cell biology.
We argue here that “the future of CAR-T will be defined as much by how we build the cells as by what they target.” By situating manufacturing platforms as active determinants of CAR-T cell biology shaping clonal fitness, epigenetic state, persistence, and long-term immune recovery, this review provides a conceptual framework that connects laboratory methods with clinical consequences.4–7 In doing so, we aim not only to synthesize existing evidence but also to chart a forward-looking roadmap for safer, more durable, and more accessible cellular immunotherapies.
CAR-T MANUFACTURING PLATFORMS
The manufacturing platform is more than a technical detail; it actively determines the genetic architecture, epigenetic state, and ultimate clinical performance of CAR-T cells. 8 Current strategies can broadly be divided into viral vector-based and nonviral approaches, each with distinct advantages, liabilities, and implications for translational scalability.
Viral vectors: The established backbone
γ-Retroviral and lentiviral vectors have dominated CAR-T development for over two decades and remain the foundation of all currently licensed products.4,8 Their principal strength lies in stable integration of the CAR transgene into the host genome, ensuring durable expression and sustained antitumor activity. 8 Robust transduction efficiencies and extensive preclinical safety data have provided regulatory confidence, enabling multiple approvals and widespread clinical adoption. 4
There are, however, meaningful biological and practical distinctions between the two major viral platforms. γ-Retroviruses preferentially integrate near transcription start sites and enhancer regions, raising concerns about insertional mutagenesis, although thus far these risks have not translated into clear clinical events in CAR-T settings. 8 Lentiviruses, derived from HIV, exhibit broader integration patterns and can efficiently transduce nondividing T cells, which may contribute to a more favorable safety and functional profile.4,8 These features, combined with decades of optimization, explain why lentiviral vectors have become the preferred viral platform in most contemporary products.
Despite these advantages, viral vectors carry significant liabilities. The production of GMP-grade viral stocks is costly, technically complex, and highly centralized, creating long turnaround times and recurrent global supply bottlenecks. 4 Integration remains semirandom, with a residual risk of clonal dominance that mandates long-term follow-up. 8 Moreover, the limited cargo capacity of viral vectors constrains the incorporation of multigene constructs such as switch receptors, safety switches, or complex synthetic circuits. 8 These constraints have catalyzed the search for nonviral alternatives.
Nonviral systems: Expanding opportunities
A new wave of nonviral platforms has emerged to address these limitations. Transposon systems, such as Sleeping Beauty and PiggyBac, enable stable genomic integration at lower cost and with higher cargo capacity than viral vectors.4,11 The Sleeping Beauty system, engineered from a fish transposon, has advanced into several early-phase clinical trials and demonstrated feasibility with manageable toxicity.9,14 PiggyBac, derived from insects, offers even larger cargo capacity but has raised safety concerns following reports of CAR-T-derived lymphomas driven by clonal dominance. 11 These findings highlight the promise and peril of transposons: scalable and inexpensive, yet in need of more rigorous mechanistic characterization and improved control over integration profiles.
CRISPR/Cas9-based strategies add a new dimension by enabling site-specific knock-in of CAR constructs into “safe harbor” loci such as T-cell receptor alpha constant (TRAC). This approach simultaneously disrupts the endogenous T-cell receptor, reducing the risk of graft-versus-host disease in allogeneic settings, and standardizes CAR expression levels, which may improve product consistency. 10 Several first-in-human trials have shown feasibility, though challenges remain with off-target editing, chromosomal translocations, and the logistics of scaling such precision editing for commercial use. 10
mRNA-based CAR-T represents a transient, nonintegrating alternative. Electroporation of CAR mRNA generates cells with short-lived expression, typically lasting only a few days. 12 While insufficient for durable remissions, this feature can be advantageous in specific clinical contexts. Transient CAR-T has been explored as a bridging therapy before hematopoietic stem cell transplantation, as a safety-testing platform for novel CAR constructs, and as a potential repeat-dosing strategy to mitigate long-term toxicities. 12 Several academic centers and companies are now piloting mRNA CAR-T pipelines, reflecting growing recognition that transient platforms may complement, rather than compete with, durable viral or transposon-based approaches.
Emerging nanotechnology-enabled approaches are also being explored as nonviral alternatives for CAR engineering. Lipid nanoparticles, polymeric nanocarriers, and biomimetic nanosystems can deliver CAR-encoding mRNA or gene-editing cargo directly into T cells, potentially enabling transient or even in vivo CAR-T generation while reducing the risks associated with integrating viral vectors.15,16 In parallel, engineered exosomes and hybrid nanovesicles have demonstrated the ability to transfer cytotoxic proteins, CRISPR/Cas systems, and immunomodulatory molecules with lower immunogenicity and reduced risk of uncontrolled cellular proliferation, supporting their potential as complementary cell-free immunotherapeutic platforms.15,16
Platform-dependent cell states
Beyond the mechanics of gene delivery, emerging evidence suggests that each manufacturing platform leaves a distinct biological imprint on the resulting CAR-T product. 7 Viral transduction imposes cellular stress responses and creates heterogeneous integration-driven clonal diversity. Electroporation-based approaches can induce DNA damage responses and distinct transcriptional states, while CRISPR-mediated editing engages endogenous DNA repair machinery with consequences for cell-cycle progression and epigenetic remodeling. 7 Collectively, these “platform-dependent cell states” may preconfigure persistence, exhaustion, and clonal fitness even before infusion, underscoring that the choice of manufacturing platform is not neutral; it is a determinant of therapeutic biology.
These contrasts can be distilled into a schematic comparison of viral and nonviral platforms, illustrating how vector biology imprints integration patterns, cellular stress, and ultimately CAR-T phenotype (Fig. 1).
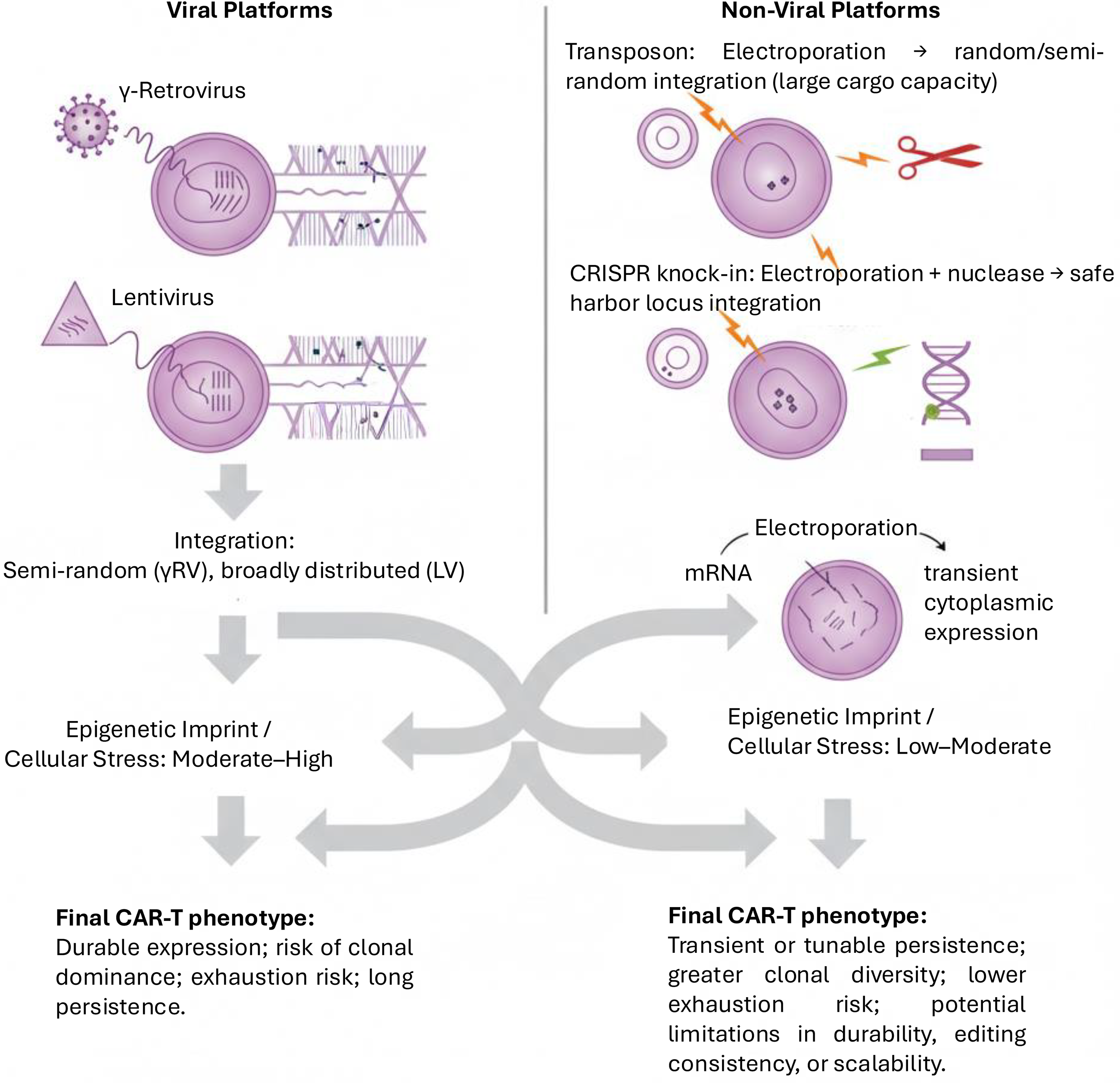
Viral versus nonviral CAR-T manufacturing platforms. Viral vectors (γ-retrovirus, lentivirus) achieve stable integration but impose higher genomic stress, leading to durable expression with risks of clonal dominance, exhaustion, and long persistence. Nonviral approaches (transposons, CRISPR knock-in, mRNA) enable larger or transient payloads with lower-to-moderate cellular stress, producing products with tunable persistence, greater clonal diversity, and lower exhaustion risk. The choice of platform imprints integration biology, epigenetic state, and final CAR-T phenotype. CAR, chimeric antigen receptor; mRNA, messenger RNA.
CLINICAL OUTCOMES AND CAVEATS
The clinical performance of CAR-T products reflects not only antigen target and patient population but also the manufacturing platform used to generate the cells. Pivotal viral-vector programs now provide long-term follow-up that defines the current benchmark, while nonviral approaches are beginning to yield informative early signals. Interpretation across studies requires caution, as eligibility criteria, use of bridging therapies, conditioning regimens, and endpoint definitions vary widely, limiting cross-trial comparability.4,5
Viral vector–based CAR-T (long-term benchmarks)
Viral vectors remain the backbone of all licensed CAR-T therapies, with extended follow-up confirming that a subset of patients can achieve durable remissions and, in some cases, long-term survival. Trials such as ZUMA-1 with axicabtagene ciloleucel, JULIET with tisagenlecleucel, and TRANSCEND with lisocabtagene maraleucel have established the therapeutic potential of viral platforms in relapsed or refractory large B-cell lymphoma.3,17,18 In multiple myeloma, both ciltacabtagene autoleucel and idecabtagene vicleucel have demonstrated clinically meaningful and durable activity in heavily pretreated populations. 19 Collectively, viral programs define efficacy and safety standards, marked by high initial response rates, a subset of long-term survivors, and well-recognized toxicities, including cytokine release syndrome, neurotoxicity, and chronic sequelae such as B-cell aplasia.3–5,20
Nonviral CAR-T (emerging signals and cautionary lessons)
Nonviral systems are now entering the clinic with instructive lessons. Sleeping Beauty–engineered CAR-T cells have demonstrated feasibility and manageable safety in early-phase studies, although efficacy has so far been less robust than viral comparators.9,21 PiggyBac offers large cargo capacity but has raised safety concerns after reports of product-derived lymphomas, reframing the discussion of risk.11,22,23 CRISPR/Cas9-mediated knock-in has reached first-in-human testing, enabling precise integration and the development of allogeneic “off-the-shelf” products, but concerns remain regarding off-target events and genomic rearrangements.10,24,25 mRNA CAR-T represents a transient, nonintegrating approach that is being investigated for bridging, repeat dosing, and rapid prototyping rather than long-term disease control.12,26
Cross-trial synthesis
Direct comparisons between viral and nonviral platforms remain heuristic rather than definitive. Viral CAR-T programs benefit from large-scale, multicenter trials with mature follow-up, whereas nonviral approaches remain early and heterogeneous. Variability in trial design further complicates interpretation. Nevertheless, clear patterns are emerging: viral vectors set the benchmark for efficacy and durability, while nonviral systems provide opportunities for scalability and innovation but introduce distinct platform-specific risks that require mechanistic vigilance (Table 1).
Clinical outcomes from pivotal chimeric antigen receptor–T trials
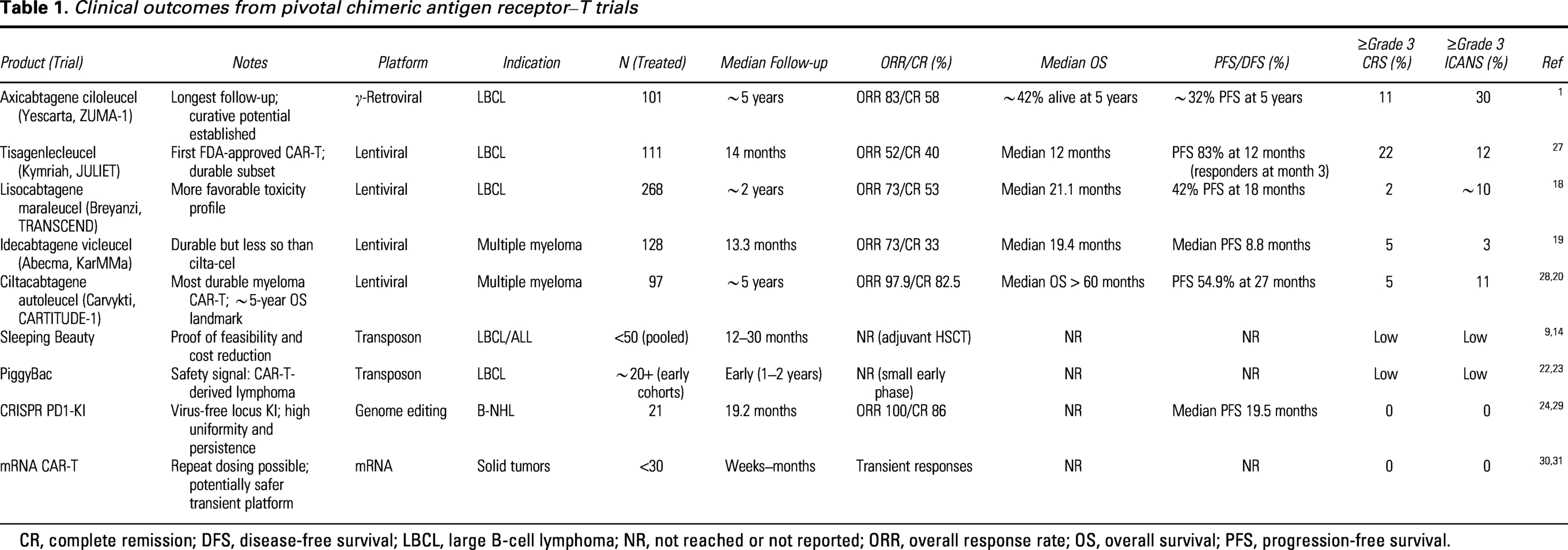
CR, complete remission; DFS, disease-free survival; LBCL, large B-cell lymphoma; NR, not reached or not reported; ORR, overall response rate; OS, overall survival; PFS, progression-free survival.
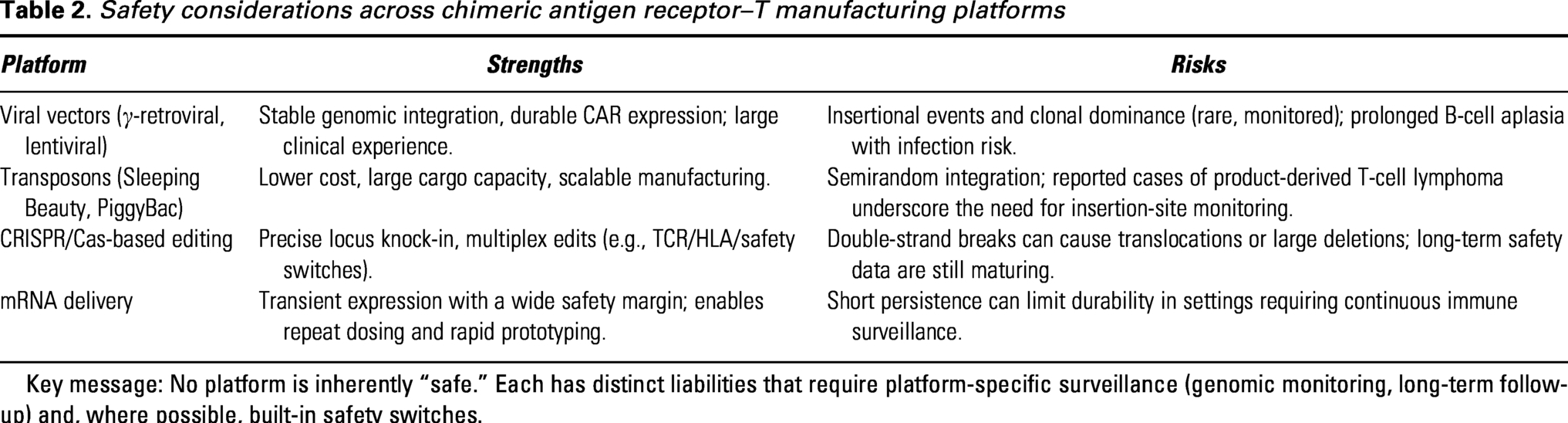
Key platform-specific safety considerations are summarized in Table 2.
Safety considerations across chimeric antigen receptor–T manufacturing platforms
Key message: No platform is inherently “safe.” Each has distinct liabilities that require platform-specific surveillance (genomic monitoring, long-term follow-up) and, where possible, built-in safety switches.
MECHANISTIC INSIGHTS BEYOND INTEGRATION
The biological consequences of manufacturing choices extend far beyond transgene delivery. Each platform interacts with T cells in ways that shape genomic stability, clonal composition, epigenetic state, and ultimately therapeutic fate. These influences are not incidental; they establish the biological baseline from which efficacy, persistence, and safety emerge. Understanding these mechanisms is therefore essential for interpreting variability in clinical outcomes and for designing safer, more effective CAR-T therapies.32,33
Integration biology: from risk to precision
Stable CAR expression has traditionally relied on genomic integration, but the precise mode of integration carries distinct biological and regulatory consequences. γ-Retroviral vectors preferentially integrate near transcription start sites and enhancer-rich regions, whereas lentiviral vectors distribute more broadly with reduced proto-oncogene bias.32,33 Transposon systems such as Sleeping Beauty and PiggyBac can deliver large payloads, but clinical observations of PiggyBac-associated T-cell lymphomas have reframed integration biology as a central regulatory concern requiring insertion-site monitoring.9,11,22 In contrast, CRISPR/Cas9 knock-in enables targeted integration at “safe-harbor” loci (e.g., TRAC or PD-1), standardizing expression while introducing double-strand-break liabilities (structural variants, translocations, off-targets) that necessitate dedicated long-term surveillance.10,13,24
Clonal fitness: Manufacturing as a selection event
The infusion product is a mosaic of clones with variable proliferative capacity, metabolic flexibility, and resistance to exhaustion; manufacturing acts as the selective bottleneck. Culture duration, activation strength, cytokine cocktails, and delivery modality determine which clones dominate. Protocols centered on IL-2 and strong activation often enrich short-lived effector phenotypes, whereas IL-7/IL-15-supported processes tend to preserve memory-like subsets with improved persistence.7,33,34 Multiomic profiling (T-cell receptor [TCR] clonotyping, mitochondrial stress testing, single-cell RNA/ATAC-seq) distinguishes “fit” stem-like programs associated with durable control from exhaustion-skewed programs that portend relapse.35–37
Epigenomic imprinting: The hidden memory of manufacturing
Beyond clonal composition, manufacturing imprints durable chromatin states that preconfigure in vivo trajectories. Viral transduction can trigger DNA-damage responses; electroporation induces transcriptional stress; cytokine milieu steers chromatin toward stemness (e.g., TCF7/LEF1) or exhaustion (e.g., TOX/NR4A). Single-cell studies show that TCF7+ memory-like programs predict persistence, whereas TOX- and NR4A-driven programs foreshadow functional decline.
Synthetic biology convergence
Platform choice constrains which next-generation designs are feasible. Armored or logic-gated constructs and multigene circuits often exceed viral cargo limits, favoring transposons or CRISPR knock-ins; conversely, switchable designs may benefit from transient mRNA delivery.10,33 As circuit complexity moves into the clinic, platform-design compatibility becomes a determinant of feasibility, monitoring, and standardization. 33
Mechanistic framework
Manufacturing imprints therapeutic products across three interdependent layers: integration biology, which dictates genomic stability; clonal fitness, which governs persistence versus exhaustion; and epigenomic imprinting, which sets long-term programming. Synthetic biology overlays this framework with new constraints and opportunities, enabling manufacturing strategies to more precisely shape CAR-T persistence, functional programming, and safety profiles.
These three interdependent layers, genomic integration, clonal fitness, and epigenomic imprinting, can be synthesized into a mechanistic framework that links manufacturing decisions to long-term CAR-T outcomes (Fig. 2).
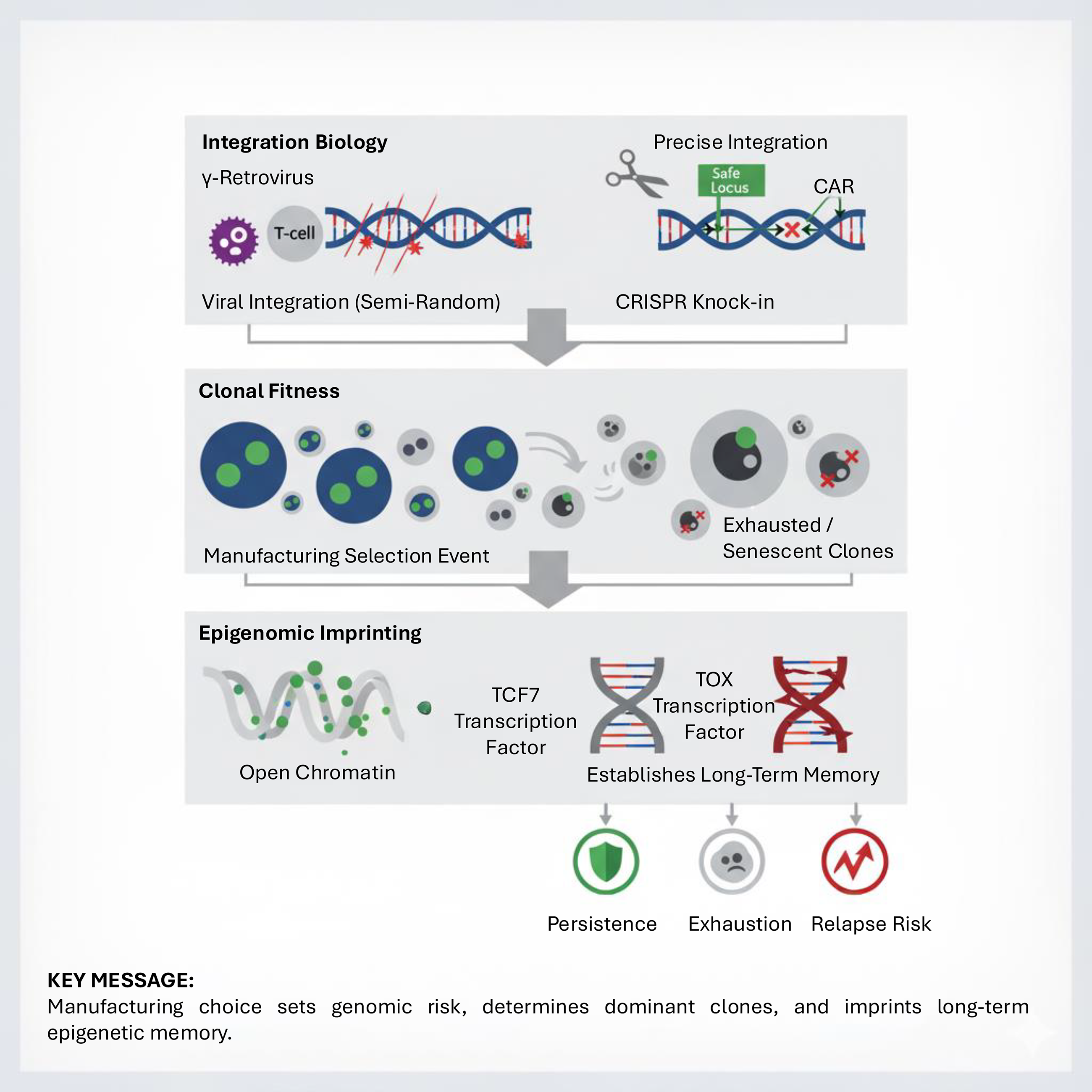
Mechanistic framework of manufacturing: integration biology, clonal fitness, and epigenomic imprinting. Manufacturing leaves a biological imprint across three mechanistic layers. Integration biology determines genomic stability, with viral vectors integrating semirandomly and CRISPR knock-ins enabling precise insertion into safe loci. Clonal fitness reflects manufacturing as a selective bottleneck, enriching for clones with proliferative dominance while others become exhausted or senescent. Epigenomic imprinting programs long-term fate, with TCF7-associated memory chromatin states predicting persistence and TOX-driven states foreshadowing exhaustion and relapse risk. Together, these layers frame manufacturing as a determinant of therapeutic destiny.
IMMUNE RECOVERY AND LONG-TERM SEQUELAE
Beyond immediate efficacy and acute toxicities, CAR-T therapy induces durable perturbations in immune homeostasis that are shaped by the manufacturing platform. Gene-transfer modality, culture conditions, and expansion kinetics not only determine antitumor activity but also imprint the pace and quality of immune reconstitution after infusion. Differences in B-cell and T-cell recovery trajectories frame a central clinical tension: sustaining tumor surveillance without entrenching immunodeficiency.4,33,38
B-cell aplasia: Biomarker, burden, and management
For CD19- and B-cell maturation antigen-directed products, persistent B-cell aplasia with hypogammaglobulinemia serves as a pharmacodynamic surrogate of CAR activity and correlates with durable disease control.4,39 Yet, this same persistence drives infection risk and necessitates long-term IgG replacement. Platform choice influences this trajectory: stable integrating systems (γ-retroviral, lentiviral, and some transposons) typically prolong aplasia, whereas mRNA-based CAR-T induces only short-lived depletion and may suit contexts where durable immunosuppression is undesirable, such as bridging to HSCT. Emerging reports suggest that some transposon products allow earlier recovery, but PiggyBac safety signals mandate careful interpretation.10,11 Mitigation strategies include routine IgG monitoring, individualized replacement, vaccination planning once B-cell and IgG recovery permit, and incorporation of controllable or suicide switches for products at risk of protracted aplasia.
B-cell aplasia is both a biomarker and a burden. The challenge is not simply managing hypogammaglobulinemia but learning to titrate persistence to balance tumor control against infection liability.
T-cell reconstitution: Clonal dynamics, determinants, and consequences
T-cell recovery after CAR-T is highly heterogeneous and reflects how the product was built. Extended culture with IL-2 and strong activation often generates effector-biased products that contract quickly, delaying CD4/CD8 reconstitution. By contrast, approaches preserving stem-like pools (IL-7/IL-15 support, shortened culture, or site-specific knock-in) yield more balanced and durable repopulation.7,33 Recovery is also influenced by lymphodepletion intensity, prior therapies, and intercurrent infections. Clinically, delayed reconstitution manifests as poor vaccine responses and opportunistic viral or fungal infections. In rare instances, persistent clonal skewing or genomic instability raises concern for secondary lymphoid malignancies, including CAR-T-associated T-cell lymphomas. While the clearest signals to date involve PiggyBac transposon products, 11 vigilance across all integrating platforms is warranted. 40
T-cell reconstitution is not a passive rebound; it is a mirror of how the cells were manufactured. The phenotype encoded at the factory dictates the immune landscape for months to years after infusion.
Infections as the leading nonrelapse mortality signal
Across long-term cohorts, infections have overtaken cytokine storm as the leading cause of nonrelapse mortality. Early bacterial events dominate in the neutropenic window, while later phases are characterized by reactivation of DNA viruses (HSV, CMV, BK), respiratory pathogens, and invasive fungi.4,38,41 This epidemiology links therapeutic persistence to clinical hazard: the very durability that predicts remission also sustains immunodeficiency. Standard management includes antibacterial prophylaxis during neutropenia, PJP prophylaxis for at least 6–12 months (longer if CD4 <200/µL), HSV/VZV prophylaxis, and center-specific CMV surveillance. Vaccination schedules should be reinitiated once sufficient B- and T-cell recovery occurs to support responses.38,41
Infection risk is not merely a side effect but the shadow of success: persistence is a double-edged sword that requires equally persistent surveillance.
Hematopoietic reserve at apheresis as a predictor
Hematopoietic reserve at the time of collection is an increasingly recognized predictor of downstream immune recovery. Low reserve reflected by unfavorable baseline counts and CD3+ yield at leukapheresis associates with prolonged post-CAR-T cytopenias, delayed T-cell reconstitution, and higher infection burden.35,42,43 Because reserve and product phenotype are interdependent, manufacturing strategies that preserve less-differentiated T-cell states may partially mitigate risk in marginal patients. Incorporating reserve metrics into upfront planning can guide intensity of prophylaxis, frequency of IgG monitoring, and timing of revaccination.
Hematopoietic reserve is the hidden denominator of immune recovery. Without factoring in marrow fitness at collection, platform comparisons risk confounding biology with baseline fragility.
Platform-dependent trajectories and management blueprint
Taken together, vector choice and process parameters help shape immune trajectories. Viral vectors and many transposon products yield prolonged B-cell aplasia and slower T-cell rebound; mRNA CAR-T provides transient depletion with faster recovery; and CRISPR knock-in strategies that conserve memory-like phenotypes may balance persistence with physiological repopulation, though long-term data are still maturing.7,10,33 A pragmatic blueprint is to pair platform-aware surveillance (longitudinal IgG, lymphocyte subsets, pathogen monitoring) with tiered prophylaxis and vaccination and to consider circuit-level controls, drug-gated or suicide switches, where persistent aplasia or recurrent infections threaten quality of life.
The future of immune recovery management will be platform-aware and programmable. The same design principles that dictate clonal fate may eventually enable programmable “immune rheostats” capable of tuning CAR persistence, immune recovery, and toxicity management according to disease context and patient-specific risk.
DEMOCRATIZATION OF CELL THERAPY
The transformative efficacy of CAR-T therapy has been counterbalanced by prohibitive cost, limited access, and a highly centralized manufacturing model. Licensed viral vector-based CAR-T products in the United States and Europe carry list prices in the range of USD 400,000–500,000 per infusion, excluding hospitalization and supportive care.44,45 Reliance on GMP-grade viral vectors, complex logistics, and prolonged vein-to-vein times has created a therapeutic landscape in which access is confined to a small subset of patients in high-income countries. This imbalance underscores an urgent need for strategies to democratize cell therapy, reducing cost, broadening access, and ensuring global scalability without compromising quality. 4
Cost reduction and manufacturing efficiency
Nonviral platforms offer a promising route to cost containment. Transposon systems, requiring only plasmid DNA and electroporation, bypass the capital-intensive processes of viral vector production. 21 mRNA CAR-T cells can be generated within days at relatively low cost, supporting repeat-dosing paradigms. CRISPR-based knock-in remains resource-heavy, but efficiencies in editing, automation, and reagent supply chains may eventually render it cost-competitive. The central caveat is that economic gains must not compromise quality: manufacturing shortcuts that reduce batch-to-batch consistency would undermine clinical credibility and jeopardize regulatory approval. 46
Cost discussions should not be framed narrowly as “cheap versus expensive.” The deeper question is whether nonviral systems can deliver credible cost savings without eroding the reproducibility and trustworthiness that underpin therapeutic adoption.
Accessibility in low- and middle-income countries
The inequity in CAR-T access is stark. While thousands of patients have been treated in North America, Europe, and China, most of the global population remains excluded. 47 For countries such as India and Vietnam, the high cost of viral vectors and the absence of centralized GMP facilities are prohibitive barriers. Transposon- and mRNA-based platforms, deployable at lower cost and with more flexible infrastructure, are being piloted as feasible solutions. Success, however, will depend on partnerships between academic centers, local biotech companies, and public health systems to adapt CAR-T manufacturing to regional contexts.
For LMICs, the “vector problem” is really an infrastructure problem. Without creative public–private models, even cheaper vectors will not translate into access.
Decentralized, point-of-care models
Decentralized, point-of-care (POC) manufacturing has emerged as one of the most promising transformative opportunities. Early experiences in China show that hospital-based CAR-T production can cut vein-to-vein times from weeks to days while reducing costs. 48 Closed and automated platforms further enhance feasibility and safety, enabling smaller centers to deliver advanced therapies without industrial-scale infrastructure. Nonviral approaches are particularly compatible with POC models, since they bypass viral supply chains and support rapid iteration.
POC manufacturing is not just about speed; it represents a paradigm shift from product to process. The therapy becomes a locally produced service, raising new questions about standardization and oversight.
Regulatory harmonization and global quality
The globalization of CAR-T raises complex regulatory challenges. Viral vector-based products are tightly regulated in the United States and Europe, but harmonization across regions remains incomplete. For nonviral and decentralized models, new standards are urgently needed to define acceptable integration-site profiles, off-target thresholds, and release criteria. Without harmonization, disparities in rigor risk undermining public trust. Collaborative initiatives involving the FDA, EMA, NMPA, and regulatory bodies from emerging markets will be required to establish global benchmarks. 49
Regulatory harmonization is not simply bureaucratic alignment; it is the currency of global trust. Without it, democratization risks devolving into a two-tiered system where “cheap CAR-T” is viewed with suspicion.
Toward true democratization
Democratizing CAR-T therapy requires more than technical innovation; it demands a reimagining of the delivery ecosystem. Nonviral platforms, decentralized manufacturing, and regulatory alignment together outline a roadmap toward equitable access. The ultimate test of democratization will not be whether CAR-T remains transformative in wealthy health systems, but whether its benefits can extend to the majority of patients worldwide.
The central question is not “Can CAR-T cure cancer?” We know it can. The question is “Who gets cured?” Democratization is the axis on which the future relevance of CAR-T will turn.
FUTURE ROADMAP
The rapid evolution of CAR-T therapy suggests that the next decade will be defined less by incremental changes and more by strategic convergence, integrating manufacturing innovation with synthetic biology, disease-specific tailoring, and global accessibility. The roadmap ahead is multidimensional, aligning technological potential with clinical and societal imperatives. 33
Hybrid strategies and bridging approaches
Rather than a binary choice between viral and nonviral platforms, hybrid models are poised to dominate. Viral vectors will likely remain a reliable option for durable integration in settings where long-term surveillance is essential, while site-specific CRISPR knock-in can add precision at defined loci to standardize expression and reduce insertional randomness.10,50 Transposons bring large cargo capacity that enables armored or circuit-rich designs and can be layered with viral or editing approaches when complexity demands it. 33 In parallel, mRNA CAR-T is well-suited for bridging or first-in-human safety prototyping, providing transient control while more durable products are prepared, or derisking novel constructs before committing to stable integration. 33
The winning strategies will treat platforms as composable parts, not mutually exclusive choices.
Allogeneic and “off-the-shelf” CAR-T
Transitioning from autologous to allogeneic products hinges on precise TCR disruption and immune-evasion edits (e.g., human leukocyte antigen engineering), which are most tractable with modern genome editing (CRISPR, base editing).51–54 Proof-of-concept human studies with multiplex CRISPR editing and early clinical experiences with gene-edited allogeneic products have established feasibility, though questions of durability, immunogenicity, viral reactivation risk, and regulatory complexity remain central.
Allogeneic progress will be paced as much by editing quality and release testing as by clinical activity.
Precision matching of platform to disease context
Emerging experience supports disease-specific matchmaking. Rapidly progressive leukemias may tolerate transient or bridging expression before transplant; lymphomas requiring prolonged surveillance favor stable integration and memory-skewed phenotypes; myeloma’s marrow niche may benefit from armored payloads enabled by larger cassettes or knock-ins.4,33
Platform choice is becoming a therapeutic variable, not merely a manufacturing preference.
Increasingly, the intrinsic limitations of each manufacturing platform can be reframed as programmable design features rather than unavoidable liabilities. Durable integrating platforms may remain preferable in diseases requiring long-term immune surveillance, because CD19-directed CAR-T cells can produce prolonged remissions and may be curative in a subset of patients with B-cell lymphomas. 4 Conversely, transient platforms such as mRNA CAR-T may be intentionally deployed when reversibility and avoidance of genomic integration are prioritized, including bridging therapy, repeat dosing, early construct testing, or settings in which excessive on-target toxicity is a concern. 12 Site-specific CRISPR knock-in strategies provide a complementary route by reducing random integration and enabling more standardized CAR expression programs; preclinical and early clinical studies have shown that nonviral or virus-free targeted CAR insertion can preserve memory-like phenotypes, reduce tonic signaling or exhaustion, and support antitumor activity.10,25 In this framework, manufacturing constraints are no longer viewed solely as technical barriers, but as tunable biological variables that can be matched to disease context, toxicity tolerance, desired persistence, and immune recovery kinetics.
Synthetic biology integration
The next wave of CAR-T will be shaped by synthetic circuits logic gating, inducible switches, and programmable secretory modules. Viral vectors face cargo and modularity limits, whereas transposon and CRISPR-based systems better accommodate multigene architectures; transient mRNA complements these by enabling rapid iteration and safer early testing.50,55,56 Landmark work in TRAC knock-in, logic-gated sensing, and armored constructs has set the template for clinical translation.
Competitive advantage will shift to programs that can iterate and integrate modules quickly while meeting quality thresholds.
Convergence toward ecosystems of cell therapy
The destination is an ecosystem rather than a single “best” platform: stable viral or knock-in systems coexisting with transient mRNA; autologous lines alongside allogeneic and POC models; and circuit-enhanced constructs layered across multiple backbones.33,48 Success will depend on harmonizing these elements into scalable, safe, and globally accessible frameworks linking platform capabilities with standardized analytics and release criteria.
The challenge for the field is not only to innovate, but to integrate technically, clinically, and regulatorily.
CONCLUSION
CAR-T cell therapy has transformed the treatment landscape for hematological malignancies, yet its trajectory will be determined as much by manufacturing choices as by target selection. Viral vectors, transposons, CRISPR-based knock-ins, and mRNA platforms each bring distinct strengths and liabilities, leaving a biological imprint that influences integration biology, clonal fitness, and epigenomic imprinting, the three mechanistic layers that collectively shape therapeutic destiny. These differences translate into variable efficacy, toxicity, and accessibility, underscoring the need to view manufacturing not as logistics but as a biological determinant of outcome.
Looking forward, the convergence of diverse platforms with synthetic biology promises to expand the functional repertoire of CAR-T, enabling safer, more precise, and context-specific applications. At the same time, democratizing cell therapy through cost reduction, decentralized manufacturing, and global regulatory harmonization will be essential if the benefits of CAR-T are to extend beyond privileged health systems.
The challenge for the next decade is integration: combining technological innovation, mechanistic insight, and equitable implementation into a coherent roadmap. By explicitly recognizing and applying the three-layered mechanistic framework of genomic stability through integration biology, persistence through clonal fitness, and functional programming through epigenomic imprinting, this review establishes a conceptual foundation for the next generation of CAR-T innovation. Achieving this integration will not only define the trajectory of CAR-T but also set a paradigm for the broader field of cellular immunotherapy.
The fate of CAR-T will not hinge solely on better constructs but on whether the field can harmonize biology, engineering, and equity into a scalable and trusted therapy.
Looking ahead, several outstanding questions remain central to the evolution of CAR-T manufacturing, underscoring the uncertainties that must be addressed before true global standardization is achieved (Table 3).
Key translational questions in chimeric antigen receptor–T manufacturing
AUTHORS’ CONTRIBUTIONS
Both authors wrote the article.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable. This review synthesizes previously published studies and did not involve new studies with human participants or animals.
AVAILABILITY OF DATA AND MATERIALS
No new datasets were generated or analyzed for this article. All data discussed are from previously published sources cited in the article.
DISCLOSURE
D.-H.B. used ChatGPT (OpenAI) and Gemini (Google) as language-support tools during article preparation to assist with organization and clarity. These tools were also used to generate preliminary conceptual figure drafts based on the authors’ scientific descriptions. They were not used as sources of scientific authority. All hypotheses, interpretations, references, and conclusions were independently developed and verified by the authors, who take full responsibility for the content of the article.
AUTHOR DISCLOSURE
All authors declare no competing interests.
Footnotes
FUNDING INFORMATION
There is no funding for this article.