
Abstract

The lysosomal storage disease, mucolipidosis type II (MLII), is caused by loss of function of GlcNAc-1-phosphotransferase (GNPT), which is responsible for tagging lysosomal hydrolases with mannose 6-phosphate for delivery to the lysosome. The α and β subunits of GNPT are encoded by GNPTAB and translated as a single protein that is post-translationally cleaved in the Golgi to generate the activated α and β subunits. 1 Proteolytic cleavage of GNPT is a prerequisite for its catalytic activity. 1 One of the most frequent pathogenic variants causing MLII is a dinucleotide deletion within exon 19 of the GNPTAB gene that leads to a frameshift and a premature stop codon, 2 resulting in a truncated protein lacking the C-terminal 89 amino acids that includes half of the Stealth domain 4 within the enzyme’s catalytic core. 3 It has been proposed that antisense oligonucleotide-mediated skipping of exon 19, which removes the 56 amino acids encoded by exon 19 but keeps the rest of the reading frame intact, could potentially restore partial activity to GNPT and help mitigate the severity of the disease in patients with this mutation. 4 However, the validity of this idea has not been tested at the protein level.
In this study, we have generated GNPTAB constructs that are either wild-type or lack exon 19, and a construct termed S1S3 (with or without the nucleotides corresponding to exon 19) that encodes primarily the catalytic core of the protein and gives rise to a hyperactivated enzyme, 5 as described under experimental design below. Immunoblot analysis shows full-length GNPT to be efficiently cleaved to generate the activated β subunit of the enzyme (Fig. 1A). The activated α subunit is not detected since the V5-epitope tag is appended to the C-terminus of the protein. In addition, some uncleaved inactive αβ precursor is also observed. In contrast, GNPTΔExon 19 produces only the inactive αβ precursor and virtually no cleaved and activated β. Since the cleavage site is well upstream of exon 19, this suggests that deletion of the amino acids encoded by exon 19 precludes cleavage and activation of GNPT. In agreement with the immunoblot data, GNPTΔExon 19 enzyme activity was reduced by 99% relative to full-length GNPT (Fig. 1B). The result raises the question of whether the absence of amino acids encoded by exon 19 merely prevents proteolytic cleavage of GNPT and hence the lack of activity or if it was directly impacting the catalytic function of the enzyme.
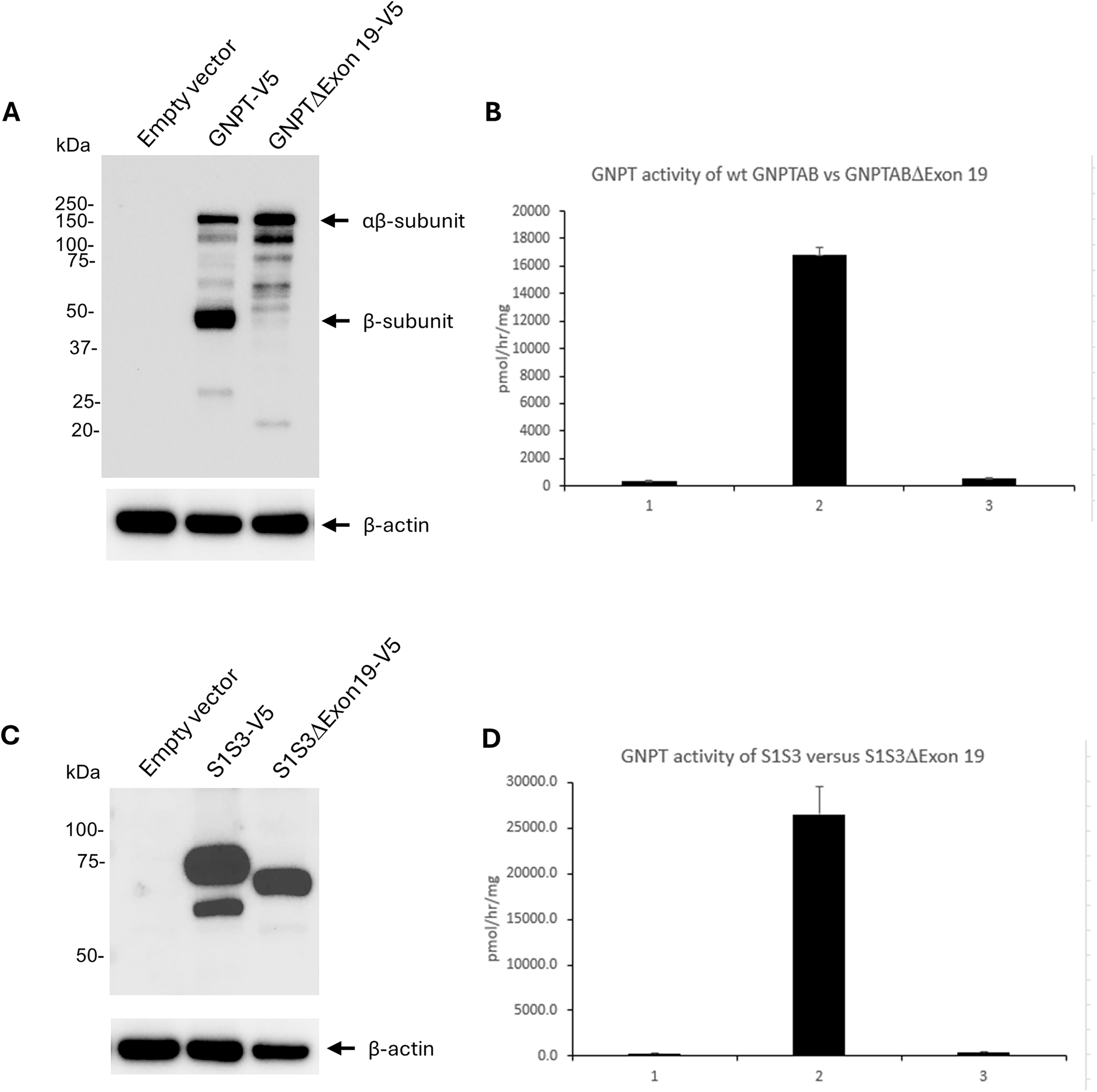
Deletion of amino acids encoded by exon 19 of the GNPTAB gene.
To circumvent the requirement for cleavage and activation, we tested the consequence of exon 19 deletion in S1S3, which is highly active despite not undergoing proteolytic cleavage into the α and β subunits. 5 While S1S3ΔExon19 is expressed reasonably well, as determined by immunoblot analysis (Fig. 1C), its catalytic activity was <1% of that of S1S3 (Fig. 1D). This result shows that removal of exon 19 amino acids directly affects the catalytic activity of the protein. Taken together, our data demonstrate that the amino acids encoded by exon 19 are indispensable for GNPT function in cells.
Therapeutic strategies that would omit exon 19 of GNPTAB would be ineffective for treatment of ML II disease. Exon 19 encodes several critical residues comprising Stealth domain 4 of the protein.3,5 We demonstrate here that deletion of this crucial exon results in complete loss of GNPT activity. While this article was under review, Goncalves et al. 6 reported very similar findings showing that deletion of exon 19 amino acids fails to give rise to the active β subunit of GNPT, leading them to the same conclusion we arrived at, namely that skipping of exon 19 is not a viable strategy to treat MLII.
EXPERIMENTAL DESIGN
The full-length GNPTAB and S1S3 constructs in the vector pcDNA6/V5-His have been described (Fig. 2A).3,5 To generate the exon 19 deletion variant, a set of complementary primers (Fig. 2B) was designed to loop out the nucleotides encoding the 56 amino acids within this exon. Quick-change mutagenesis with this primer set was performed using Pfu-Ultra (Agilent Technologies) and GNPTABpcDNA6/V5-His as template, and the reactions were treated with the restriction enzyme DpnI before being transformed into E. coli XL-10 Gold competent cells (Agilent Technologies). Bacterial colonies were screened by PCR spanning exon 19, and DNA clones that were positive for GNPTABΔExon19pcDNA6/V5-His were subject to Sanger sequencing (Fig. 2C). To generate S1S3ΔExon19pcDNA6/V5-His, the EcoRV/AflII DNA fragment that includes the exon 19 deleted region from GNPTABΔExon19pcDNA6/V5-His was switched with the corresponding fragment in S1S3pcDNA6/V5-His.
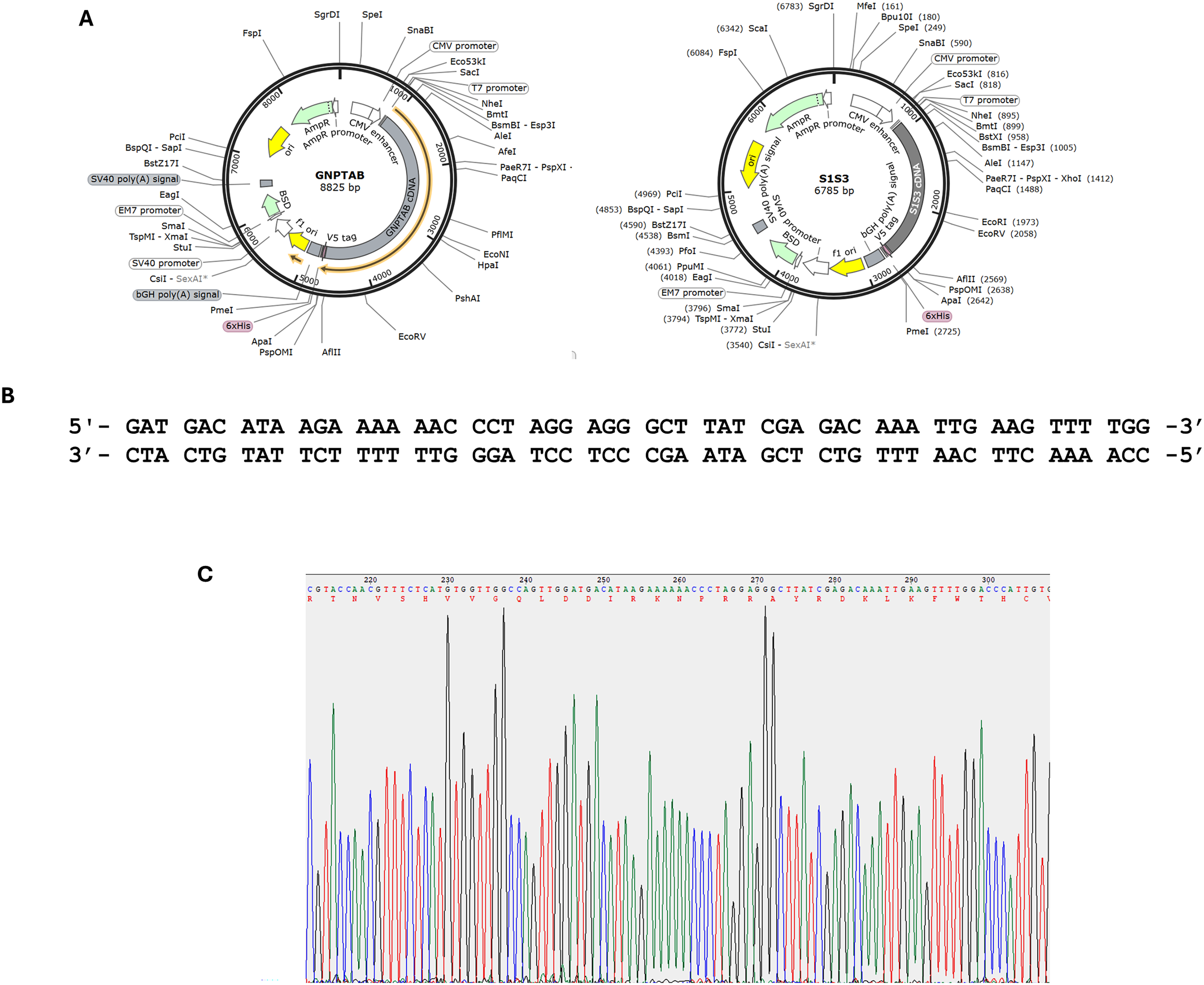
Generation of GNPTAB and S1S3 constructs lacking exon 19.
GNPTAB−/− HEK 293 T cells, 7 in a 6-well plate, were transfected with 2 µg of the indicated plasmid DNA. Cells were harvested 48 h post-transfection and processed as described in detail previously. 8 GNPT enzymatic assays were performed exactly as described in our step-by-step published protocol. 8
Footnotes
AUTHOR DISCLOSURE STATEMENT
No competing financial interests exist.
FUNDING INFORMATION
This study was supported by a grant from the National MPS Society to P.I.D.