
Abstract
Extracellular vesicles (EVs) have been investigated due to their natural biocompatibility and targeting capabilities. The specific approach of combining EVs with liposomes to create hybrid nanoparticles (ELNPs) for the delivery of the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas9) system for deletion of the HGF gene in stem cells, but their effectiveness in encapsulating large nucleic acids is limited due to their small size. This study aimed to knock out the HGF gene by the CRISPR/Cas9 system by ELNPs, and it was expected that the efficiency of the CRISPR/Cas9 system transfer would increase compared to the usual methods of using lipofectamine in stem cells from apical papilla (SCAPs). In this study, gRNA suitable for the HGF gene is designed first, and after insertion into the CRISPR/Cas9 vector, it enters Lipofectamine 2000. In the next step, ELNPs are prepared after collecting EVs and hybridizing them with liposomes containing CRISPR/Cas9 vector. Then, these integrated nanoparticles were presented to SCAPs, and the removal of HGF gene expression was evaluated at the level of RNA and protein. This study showed that the CRISPR/Cas9 system can be efficiently transferred to SCAP cells using ELNPs. Genomic DNA sequencing analyses of SCAP cells showed a unique pattern of mutation, highly likely mediated through EVs. Quantitative PCR and protein staining further showed a decrease in HGF gene expression in the knockout cells. Moreover, cell proliferation analysis showed a decrease in cell proliferation in KO-HGF adipose cells compared to the nonedited counterpart. In summary, this study highlights the supportive role of EVs in facilitating cell transfection and promoting a dominant DNA repair pattern, likely through an RNA-mediated mechanism, rather than the random insertions and deletions typically induced during CRISPR editing of the HGF gene in SCAPs.
Keywords
Background
Hepatocyte growth factor (HGF) is a multifunctional cytokine that causes different responses in different cells and tissues. Many evidence points to its role as a regulator of carcinogenesis and cancer invasion and metastasis. HGF was originally identified as a potent stimulatory factor for hepatocyte growth in vitro (Michalopoulos et al., 1984; Nakamura et al., 1984; Russell et al., 1984). HGF is a protein made up of a single chain containing 728 amino acids. Within this chain, there are specific segments, including 29 amino acids that signal where the molecule should go in a cell and a sequence of 25 amino acids that are part of its structure, possibly involved in stabilizing the molecule, contributing to its ability to interact with specific cellular receptors, or facilitating its activity in biological processes (Nakamura et al., 1984; Nakamura et al., 1989). Among the various growth factors, HGF is unique in having four Kringle domains, each located in the α subunit. In a study conducted by Moto et al., using several HGF deletion mutants, it was shown that these Kringle domains, especially the first and second domains, are essential for the correct biological function of the molecule. Kringle domains play a role in receptor–ligand binding. The human HGF gene is located on chromosome 7 at position q21.1 and consists of 18 exons and 17 introns (Fukuyama et al., 1991; Seki et al., 1991; Weidner et al., 1991).
The HGF gene is one of the important secreted factors by mesenchymal cells, including stem cells from apical papilla (SCAP). It seems that the HGF gene plays an important role in the growth process of vertebrates. In previous studies, adding an HGF inhibitor to the eye region of developing chick embryos prevented eye formation. The mesenchymal cells that surround the eye express the HGF protein, but the exact effect on eye development is unclear (Karamali et al., 2019).
Because periocular mesenchymal stem cells are not available, and SCAP has the same origin that expresses HGF, gene expression deletion in SCAP was performed. Reducing gene expression during the development process is one of the scientific methods of evaluating the role of proteins and effective factors in the development process. But sometimes the role of the gene in question is so key and important that removing the gene causes the embryo to fail, and the development process becomes difficult (Karamali et al., 2019).
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas9) is the third generation of the revolution in the history of gene editing, RNA-guided endonucleases, also known as CRISPR and Cas gene editing technologies. The CRISPR/Cas9 system has made genome engineering easier than ever with only two components, an endonuclease enzyme and gRNA, which can be modified to any desired sequence in DNA (Jinek et al., 2012). Although the CRISPR/Cas9 system is a promising gene therapy method, the CRISPR delivery into different cell lines still needs to be improved. In addition, the main obstacle in using this system is the lack of efficient delivery of the CRISPR/Cas9 system in the in vivo environment.
The CRISPR/Cas9 system has been reported mainly through viral vectors (Chong et al., 2021). On the other hand, the use of nonviral methods, such as electroporation, microinjection, and lipid nanoparticles, still have their own limitations in terms of the transfection efficiency, cell type, and in vivo applications (Du et al., 2018; Hamann et al., 2019; Meng et al., 2019). Lipofection has been widely used for in vitro and in vivo transfection applications. However, the efficiency of this approach still needs to be improved.
Extracellular vesicles (EVs) are membrane-bound particles secreted by most cell types—including cancer cells, platelets, endothelial cells, neurons, dendritic cells, and mesenchymal stem cells (Doyle and Wang, 2019; Kourembanas, 2015; Welsh et al., 2024). Notably, mesenchymal stem cells have been reported to release EVs at levels 100–1000 times higher than other cell types (Fukuta et al., 2020). EVs carry a diverse array of bioactive molecules, such as proteins, lipids, and various classes of nucleic acids, including mRNAs, microRNAs, and small interfering RNAs. However, their limited cargo capacity poses a challenge for encapsulating large nucleic acids. In particular, delivery of large constructs—such as CRISPR/Cas9 expression plasmids, which typically range from 5 to 7 kb—is inefficient due to the restricted size of EVs (Lin et al., 2018; Tian et al., 2014). This limitation underscores the need to develop alternative strategies to effectively load and deliver CRISPR/Cas9 components via EVs (Lin et al., 2018).
In this project, the goal was to assess the combinational effect of EVs with Lipofectamine 2000 on the CRISPR/Cas9 vector delivery into SCAP. We isolated EVs from three cell lines, including SCAP, adipose stem cells (ADSCs), and HEK293T cells. The CRISPR/Cas9-mediated knockout rate was measured using the analysis of Sanger sequencing data, followed by gene and protein expression analysis.
Methods
Cell culture
SCAP, ADSC, and HEK293T cells were obtained from the Royan Institute for Biotechnology. All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) with 10% fetal bovine serum (FBS) (Gibco), 1% penicillin/streptomycin (Gibco), 1% Glutamax (Gibco), and 1% nonessential amino acids (Gibco) at 37°C in a 5% CO2 environment.
Isolation of EVs
EVs were isolated from SCAP, ADSC, and HEK293T cell lines. Cells were cultured to 80% confluence, after which medium without FBS and antibiotics was added to the cell culture for 24 h. Then, the conditioned medium was collected and centrifuged for 5 min at 3000 rpm to remove particles and debris. At the next step, the EVs were isolated using the EXOCIB kit according to the protocol. For this purpose, reagent A was added to the samples at a ratio of 1:5 (v/v), vortexed for 5 min, and incubated overnight at 4°C. Then, it was centrifuged for 40 min at 3000 rpm at 4°C. The supernatant was removed, and the EV pellet was resuspended in 50 μL of reagent B and stored at −80°C for long-time use.
Determining EVs concentration using a BCA kit
The BCA kit was used to determine EV concentration. For the master stock, the BCA solution was mixed with an appropriate amount of Cu according to the protocol. A standard curve was prepared using the standard solution and master stock, with a dilution series created per protocol. Samples were then incubated in a 96-well plate at 37°C for 30 min and analyzed with a UV–visible plate reader at 562 nm. The unknown sample concentration was calculated based on the standard curve and the concentration line equation.
Constructs and gRNA
In this study, we used the pX459 and pX461 vectors, which both have the same size of 9.1 kb, encoding the Cas9 protein with either a puromycin resistance gene or EGFP gene under the GAGGS promoter/enhancer. Both plasmids also expressed the gRNA scaffold under the U6 promoter. Briefly, gRNA (5′-GTGCTGGATCTATTTTGATT-3′) was designed to target the beginning of the HGF gene, encoding the mature peptide protein using CRISPOR software (https://crispor.gi.ucsc.edu/) (Concordet and Haeussler, 2018). Synthesized gRNA was obtained from Gene Fanavaran Company (Tehran, Iran). Cloning of gRNA primers into the digested vector was performed based on the Farnham protocol (Eghbalsaied and Kues, 2023a; Guo et al., 2018). Forward and reverse gRNA strands were annealed in a 1:1 ratio with annealing buffer, followed by thermal cycling. The annealed gRNAs were diluted 1:20 with distilled water and cloned into the digested pX459 vector (50 ng) using the BbsI enzyme (Thermo Scientific) and T4 Ligase enzyme (Thermo Scientific). The cloning mixture was incubated at 16°C for 16 h. For transformation, 4 μL of the ligation mixture was added to competent Escherichia coli, incubated on ice for 30 min, heat-shocked at 42°C for 45 s, then returned to ice for 2 min. After adding 250 μL SOC medium, the mixture was incubated at 37°C for 1 h, plated on agar containing 100 μg/mL ampicillin, and incubated overnight at 37°C. Six colonies were selected, cultured in LB medium, and plasmid DNA was extracted using the QIAprep Spin Miniprep Kit. The plasmids were digested with BbsI and EcoRV enzymes (Thermo Fisher Scientific) and verified by sequencing at Genfanavaran using gRNA sequencing primers (Table 1).
Sequence of the gRNA and Primers

Production of EV–liposome hybrid nanoparticles
To create EV–liposome hybrid nanoparticles (ELNPs), EVs were incubated with Lipofectamine 2000 (Thermo Fisher Scientific). For this purpose, at first, the plasmid was diluted to 1 μg in 50 μL of Opti-MEM I Reduced Serum Medium and incubated at room temperature for 5 min, and Lipofectamine 2000 was also diluted in Opti-MEM I and combined with DNA after 5 min of incubation. EVs (4 μg/μL) were added to the plasmid–liposome complex, incubated at room temperature for 20 min, and followed by a 12-h incubation at 37°C. Transfection efficiency was evaluated using the pX461 vector containing the EGFP reporter. EGFP expression was assessed by FACS analysis. To evaluate gene editing efficiency using CRISPR/Cas9, we repeated the transfection process with the pX459 plasmid containing gRNA and puromycin genes. Transfection efficiency was evaluated using the pX461 vector containing the EGFP reporter.
Sample preparation for dynamic light dispatch test and zeta potential
The nanoparticles were prepared in the three categories of EVS, ELNP, and liposomes in the Opti-MEM culture. To prepare ELNPS, EVS, and Lipofectamine 2000, a ratio of 1:1 was mixed and then incubated at 37° C for 12 h. EV and Lipofectamine 2000 samples were added directly to the Opti-MEM culture and then measured by the Vasco Particle Size Analyzer, dynamic light dispatch test (DLS). To determine the potential of zeta, similar samples were prepared above and then determined by the Horiba-SZ100 zeta potential.
Transmission electron microscopy
EVs and ELNPs were first prepared according to Section 2.5, then stained using negative staining and examined by transmission electron microscopy.
Transfection of SCAPs
SCAPs (5 × 104 cells/cm2) were plated in 3001 plates. The pX459 plasmid with gRNA was combined with ELNPs and added to the SCAP culture medium to evaluate CRISPR/Cas9 efficiency. After 24 h, the culture medium was replaced with DMEM supplemented with puromycin (1 μg/mL). To check transfection efficiency, the pX461 plasmid was also combined with ELNPs and incubated for 24 h before replacing the medium. Cells were analyzed by flow cytometry 48 h post-transfection.
Electroporation of pX461 vector
To compare ELNP-based transfection with electroporation, we used the electroporation approach (Eghbalsaied et al., 2020; Eghbalsaied and Kues, 2023b). SCAP (106 cells) was resuspended in Opti-MEM with 1.5% Glutamax, 3% DMSO, and 20 µg of pX461 vector in a final volume of 250 μL. The electroporation mixture was transferred into a 4-mm electroporation cuvette and subjected to electroporation using the Gene Pulser Xcell Electroporation System (Bio-Rad Laboratories, Hercules, CA, USA) with a square wave protocol consisting of two pulses at 250 V, each with a pulse length of 10 milliseconds and a pulse interval of 10 s. The electrotransfected cells were then cultured in a 6-well plate, and the medium was changed the next day.
Flow cytometry analysis
After 48 h, cells transfected with the pX461 plasmid were treated with Trypsin-EDTA (0.05%) (Gibco) and resuspended in PBS. Cells were then analyzed by flow cytometry for EGFP expression, using an excitation wavelength of 480 nm and an emission wavelength of 520 nm.
DNA extraction and PCR of HGF gene
DNA was extracted from SCAP transfected with pX459 using the SinaPure DNA kit (EX6001). Prelysis buffer (100 μL) was mixed with 5 μL ribonuclease A, added to the cells, and incubated with 30 μL proteinase K at 55°C for 30 min. Lysis buffer (400 μL) was added, and the solution was centrifuged at 13,000 rpm. A wash buffer was applied, and an elution buffer was used to finalize DNA extraction. PCR was performed using YTA PCR Master Mix with 10 µM forward/reverse primers (Table 1) and 50 ng genomic DNA in a PCR program. PCR products were loaded on a 1% gel, and the desired band was excised and sequenced. Insertion/deletion analysis was performed using TIDE software (https://shinyapps.datacurators.nl/tide/) (Brinkman et al., 2014).
Reverse transcription polymerase chain reaction
Total RNA was extracted with TRI reagent (Sigma-Aldrich). cDNA was synthesized using a Biotech Rabbit cDNA Synthesis Kit. SYBR Green real-time PCR reactions were conducted in triplicate, normalized to GAPDH, and analyzed using the ΔΔCt method to calculate fold changes.
MTS assay
The MTS assay was used to determine the effect of HGF secreted by SCAPs on ADSC proliferation. ADSCs were seeded at 5 × 104 cells/cm2 in 96-well plates with SCAP-conditioned medium. After 3 days, the MTS assay was performed as per the manufacturer’s protocol (Promega, Madison, WI).
Immunofluorescence staining of HGF protein
SCAPs were fixed in 70% methanol for 30 min. The HGF primary antibody (Santa Cruz; sc‐13087, 1:100) was diluted in 10% (w/v) normal goat serum and incubated overnight at 4°C. Cells were then labeled with FITC‐conjugated antirabbit antibody (Sigma-Aldrich; F1262, 1:80) for 1 h at 37°C.
Statistical analysis
All experiments in this study were conducted in triplicate. Data analysis was carried out using GraphPad Prism 8 software, with statistical significance set at p ≤ 0.05 (Swift, 1997). All data were reported as the mean ± standard error of mean (mean ± SEM). Differences between group means were assessed using the t-test method.
Results
Insertion and verification of gRNA in pX459 vector
Following insertion of gRNA into the pX459 vector, the construct was digested with BbsI and EcoRV enzymes and analyzed by gel electrophoresis. The lack of digestion by BbsI, as expected with successful gRNA insertion, confirmed that the BbsI recognition site was disrupted by gRNA integration (Supplementary Figs. S1 and Figs. S2).
Extraction, quantification, and characterization of ELNPs
Schematic presentation of EV formation is depicted in Figure 1A. To create ELNPs, EVs were incubated with Lipofectamine 2000 and plasmid pX459 for 12 h at 37°C (Supplementary Fig. S3). EVs were successfully isolated from HEK293T, SCAPs, and ADSCs, with the protein concentration measured as 1511, 1493, and 1531 μg/mL, respectively. Statistical analysis revealed no significant differences in EV secretion among the three cell types (p value >0.05) (Fig. 1B).
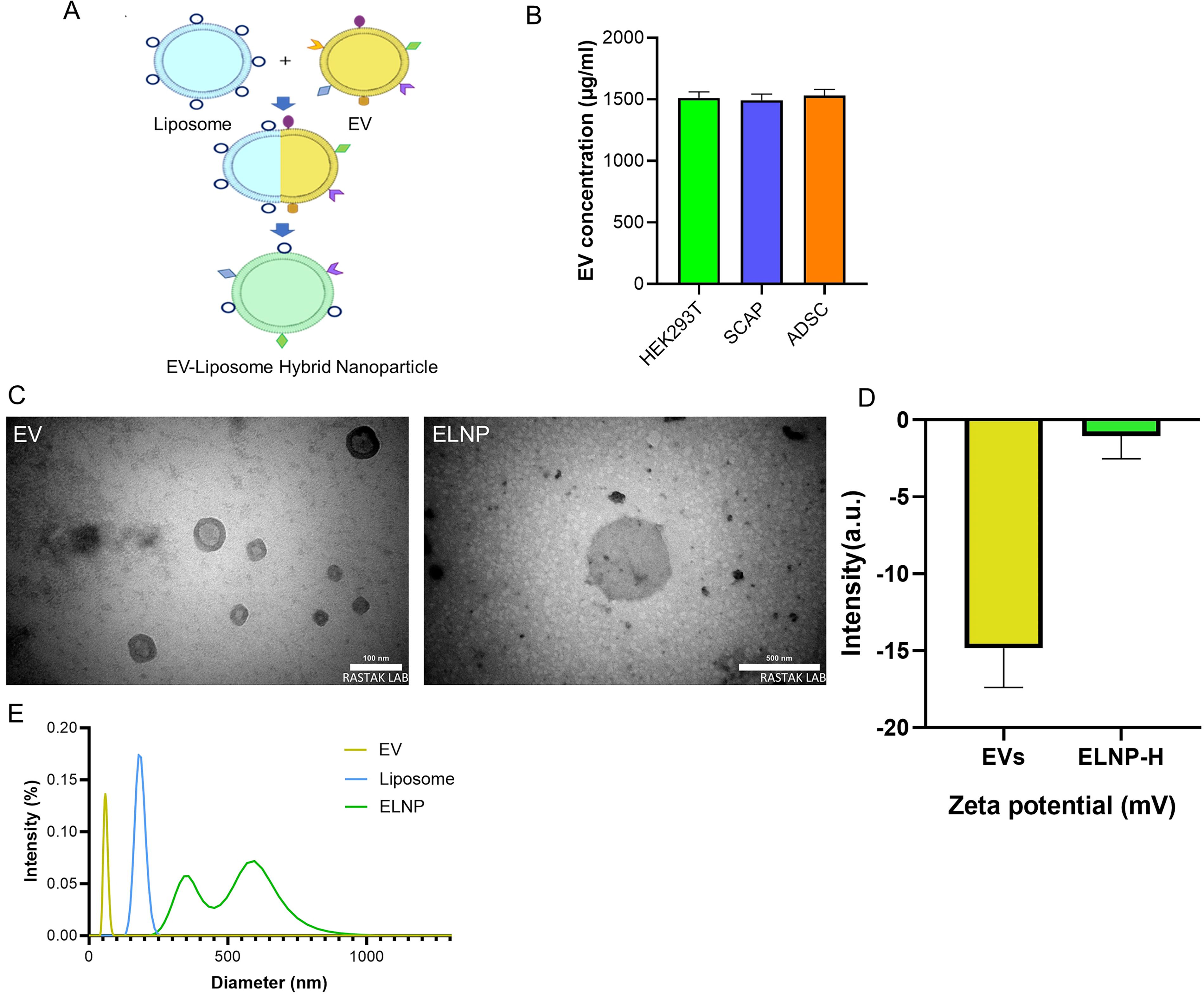
Transmission electron microscopy analysis revealed that the EVs measured ∼100 nm, while the ELNPs were around 500 nm in size, suggesting the fusion of EVs and liposomes. Zeta potential measurements indicated a value of −14.8 mV for EVs and −3.2 mV for ELNPs.
DLS analysis showed an average size of 59.35 nm (PDI: 0.008) for EVs, aligning with the expected size range for EVs. The liposomes exhibited an average size of 182.14 nm (PDI: 0.003). In contrast, the ELNPs showed two distinct size peaks at 358.28 and 595.8 nm (PDI: 049) (Fig. 1C–E). These findings indicate successful hybridization between the EVs and liposomes.
Transfection efficiency of ELNPs in SCAP cells using pX461 vector
SCAPs were treated with three ELNP groups derived from SCAP (ELNP-S), ADSC (ELNP-A), and HEK293T (ELNP-H) cells, as well as electroporation as a comparison method. Flow cytometry analysis showed that ELNP-H transfection yielded the highest efficiency at 13.3%, significantly outperforming other groups (p < 0.01) (Fig. 2). ELNP-H transfection was also significantly more efficient than lipofectamine (liposome) alone, with both transfection percentage and fluorescence intensity showing significance at p < 0.05 (Fig. 2).
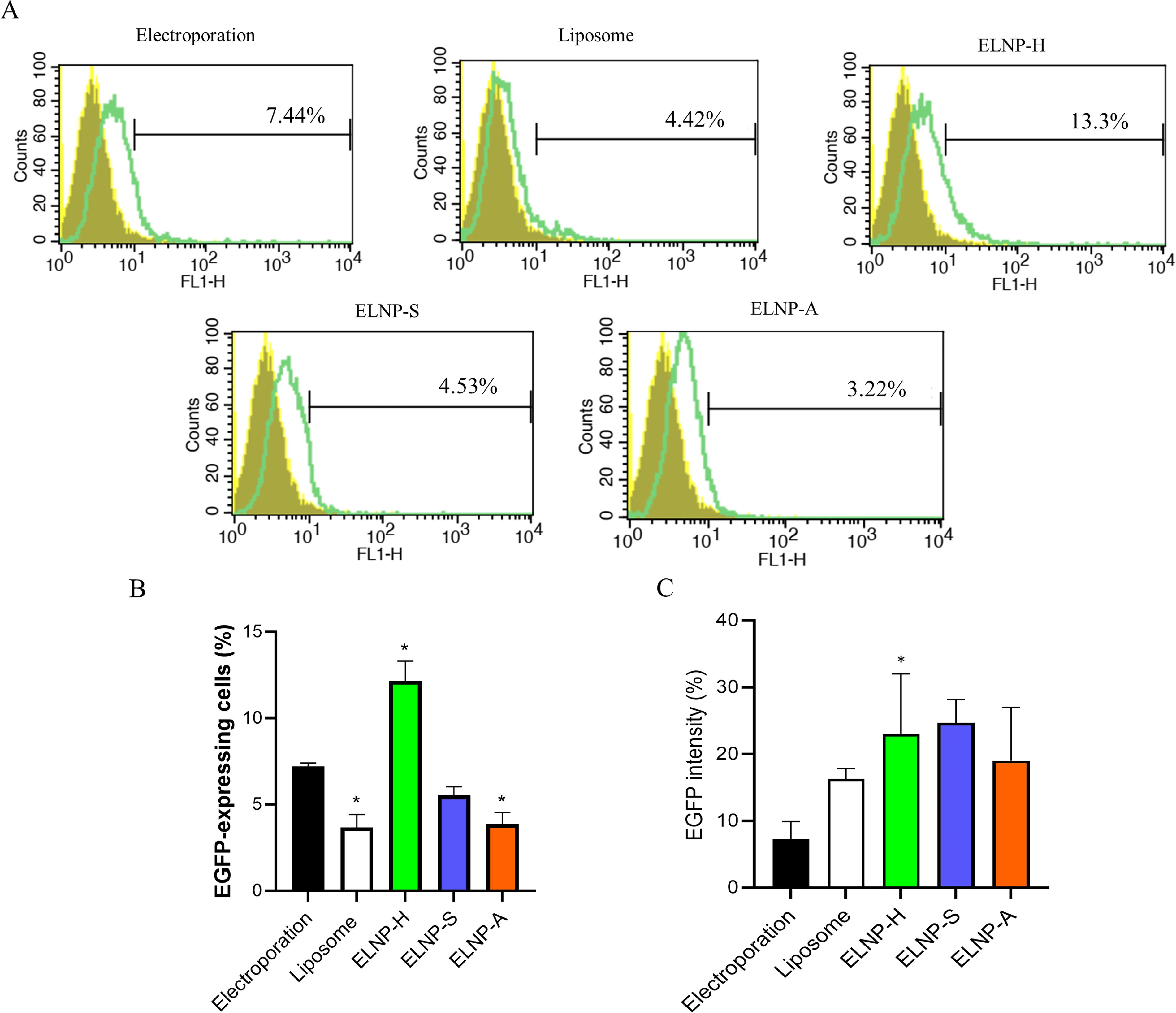
Comparison of SCAP transfection using ELNP (EV–liposome hybrid nanoparticles derived from different cells) versus electroporation.
HGF gene knockout efficiency using CRISPR/Cas9 system
CRISPR/Cas9-mediated gene editing efficiency in SCAP using ELNP group was confirmed by Sanger sequencing (Fig. 3A). Sequencing results for the knockout HGF (KO-HGF) group showed insertions at the gRNA target site, confirming successful editing (Fig. 3A and B). ICE analysis further validated the presence of a unique edited sequence with a 90% efficiency in the targeted HGF region (Fig. 3B). Further NCBI BLAST analysis of the edited sequence against the human reference RNA database revealed partial alignments with two transcripts: a long intergenic noncoding RNA and FA Complement D2 variants (Fig. 3C). These alignments suggest that endogenous RNAs with sequence homology could have potentially served as repair templates during the homologous recombination process. The identified six-nucleotide insertion results in the addition of two aspartic acid (D) residues between isoleucine at position 51 (I51) and lysine at position 52 (K52), altering the charge of the signal peptide from neutral to acidic and potentially destabilizing the protein structure (Supplementary Fig. S4).
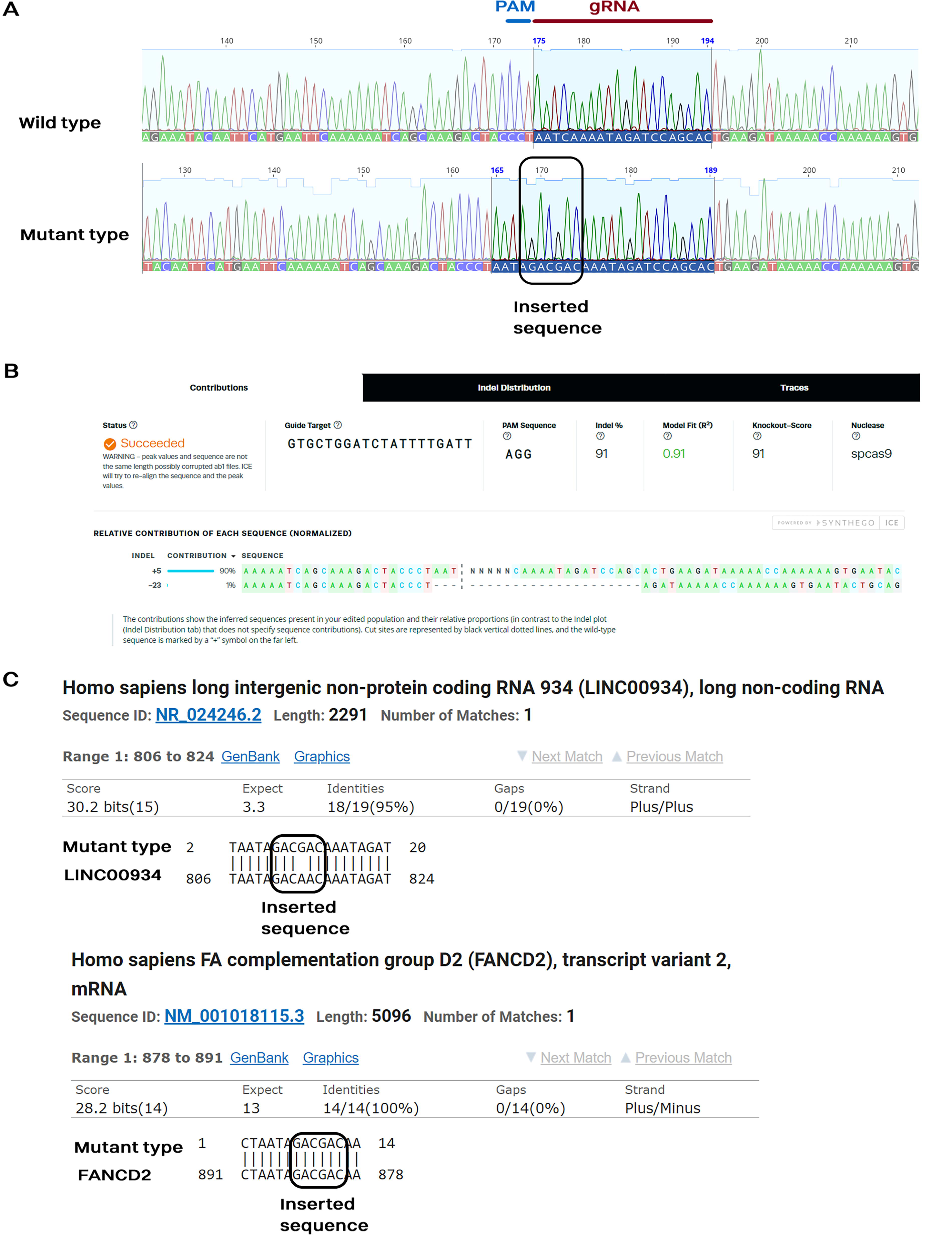
Sequencing electropherograms of PCR products and estimation of CRISPR/Cas9 editing efficiency around gRNA binding sites in ELNP compared to the control group.
Evaluation of HGF gene expression by qRT-PCR
To assess gene expression post-CRISPR/Cas9 editing, reverse transcription polymerase chain reaction (qRT-PCR) was performed on both WT and KO-HGF SCAPs. The KO-HGF group demonstrated a statistically significant decrease in HGF mRNA expression compared to the WT group (p < 0.001) (Fig. 4A).
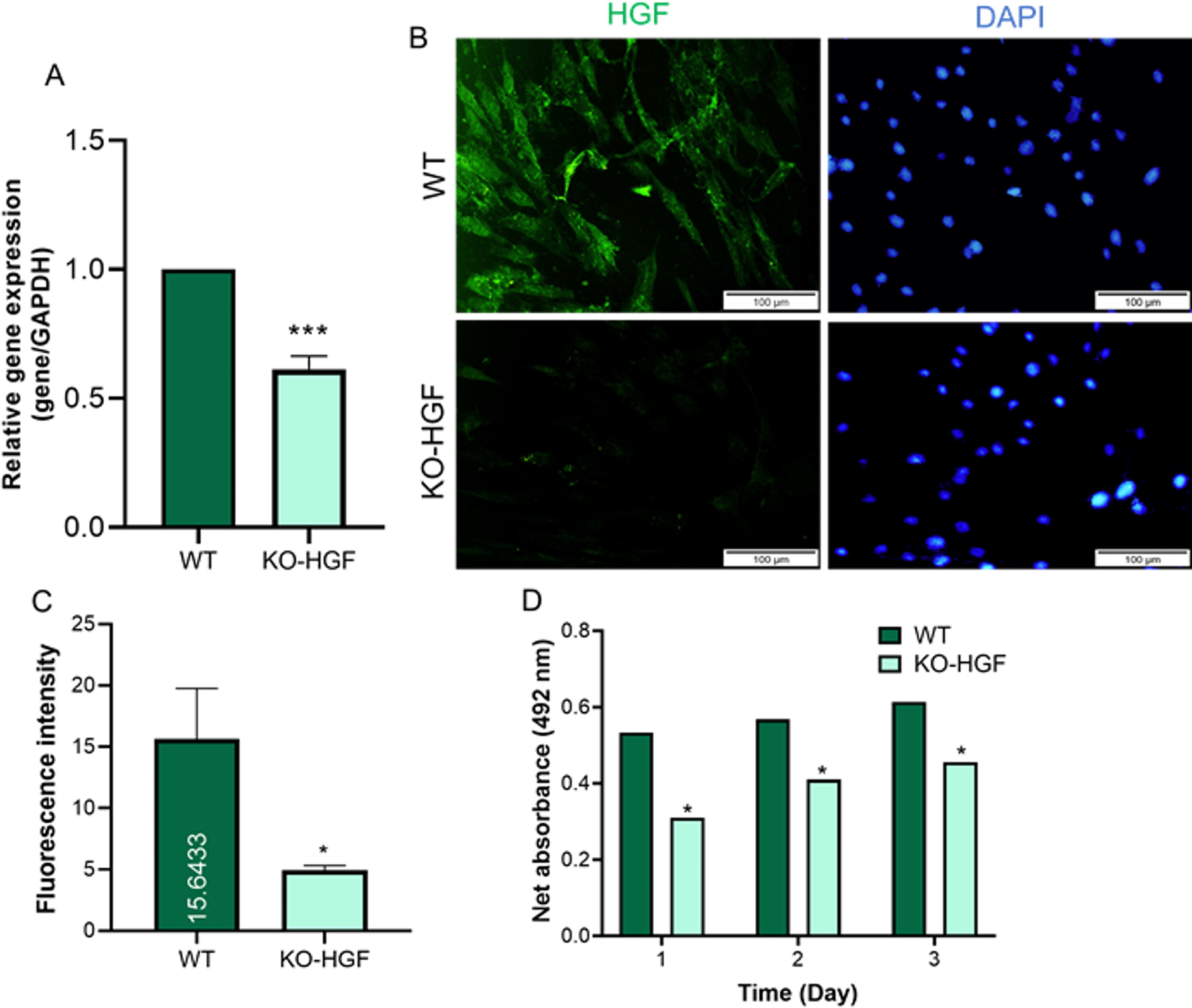
HGF expression analysis after genetic modification using the CRISPR/Cas9 editing system.
HGF protein expression analysis by immunofluorescence
Immunostaining showed a notable decrease in HGF protein levels in KO-HGF SCAPs compared to the WT group. The decrease in fluorescence intensity was quantified using ImageJ software, confirming a significant reduction in the KO-HGF group (p < 0.05) (Fig. 4B and C) (Schneider et al., 2012).
Impact of HGF knockout on cell proliferation by MTS assay
To assess the role of HGF on cell proliferation, MTS assays were conducted on ADSCs cultured in conditioned media from both WT and KO-HGF SCAPs. Over 72 h, proliferation was significantly higher in ADSCs exposed to WT-conditioned media compared to KO-HGF media (p < 0.05), indicating that reduced HGF expression diminishes SCAP’s paracrine effects on ADSC proliferation (Fig. 4D).
Discussion
Viral vectors are commonly used for gene delivery due to their high efficiency across diverse cell types. However, they are associated with drawbacks such as cytotoxicity, risk of viral infection, and complex production processes (Chen et al., 2021; Chong et al., 2021; Kim and Eberwine, 2010; Sadeghi et al., 2023). Nonviral delivery methods, including physical approaches like electroporation or microinjection, and chemical methods, offer alternatives but often cause cellular damage (Du et al., 2018; Meng et al., 2019). Among chemical strategies, liposome-based delivery is widely used due to its ability to facilitate nucleic acid transfer via lipid bilayer fusion. Despite their advantages, liposomes face challenges such as instability, poor tissue targeting, rapid clearance, and potential immunogenicity (Li et al., 2019; Mansoori et al., 2012; Shah et al., 2022). Exosomes have attracted much attention due to their natural nature, lack of cytotoxicity, and specific binding to target cells and tissues in the treatment of diseases and nanoparticle-based diagnostics (Bebelman et al., 2018; Lin et al., 2018; Théry et al., 2002). One of the cases that has received much attention is the use of hybrid nanoparticles. Recent advances have focused on hybrid nanoparticles, especially exosome–liposome nanoparticles (ELNPs), which combine the natural targeting abilities of exosomes with the stability of synthetic materials. ELNPs demonstrate high loading capacity, biocompatibility, and potential for targeted delivery of therapeutic agents, including CRISPR/Cas9 systems (Chen et al., 2021; Fu et al., 2020; Shafiei et al., 2021).
In the current study, the feasibility of delivering the CRISPR/Cas9 system using ELNPs to reduce HGF gene expression in SCAP was investigated. A major challenge in using plasmids carrying the CRISPR/Cas9 system is efficient and safe delivery to target cells, which highlights the need for a reliable and nontoxic vector. ELNPs successfully transferred CRISPR/Cas9 into SCAP cells with high efficiency and low toxicity, as no cell death was observed post-transfection. Comparatively, electroporation—a commonly used method for nonviral gene transfer—resulted in significantly lower transfection rates than ELNPs. Based on these results, ELNPs were selected as the preferred method for CRISPR delivery in this study.
In this study, two absorption peaks were observed for ELNPs. This size distribution was also observed in other papers, indicating the formation of ELNPs (Liang et al., 2022; Lin et al., 2018; Zhou et al., 2024). The presence of two absorption peaks in ELNPs is a disadvantage of this method, as it becomes impossible to separate EVs after incubation with lipid vesicles. In addition, the impairment of the biological functions of EVs due to changes in the integrity and orientation of membrane proteins is one of the disadvantages of ELNPs (Choi et al., 2021; Gorshkov et al., 2022; Li et al., 2022).
The gRNA targeting the HGF gene was strategically designed to bind exon 1, immediately downstream of the signal peptide coding region, to maximize disruption of gene function. Remarkably, ELNP-mediated delivery of the CRISPR/Cas9 plasmid resulted in a single, uniform DNA repair pattern with high efficiency (∼91%). Typically, nonhomologous end joining (NHEJ) generates heterogeneous indels following CRISPR/Cas9-induced double-strand breaks (Li et al., 2015). For comparison, our parallel experiments using electroporation to deliver CRISPR/Cas9 plasmids targeting various genes in the goat genome consistently produced multiple indel patterns (Hajian et al., 2025; Pirali et al., 2025). Although several strategies have been explored to improve editing precision, NHEJ remains the default repair mechanism in most contexts (Eghbalsaied and Kues, 2023a; Maruyama et al., 2015). The consistent repair outcome observed here following ELNP-mediated delivery and puromycin selection may therefore indicate an advantage of this system in promoting more uniform editing. EVs are increasingly recognized as carriers of diverse RNA species—including long noncoding RNAs, microRNAs, and circular RNAs—which can vary by cell type and cell cycle stage (Kim et al., 2017; Veziroglu and Mias, 2020). While the mechanism underlying the uniform repair pattern observed here remains unclear, one possibility is that RNA cargo within EVs may influence the repair environment. Recent studies have suggested that RNA molecules can participate in DNA repair processes, including as templates for homologous recombination (Durrant et al., 2024). It is also conceivable that EVs may supply nucleic acid sequences with partial homology that bias the repair machinery. However, these interpretations remain speculative and require further experimental validation. Future investigations should explore the potential contribution of EV-associated RNA or DNA in modulating DNA repair fidelity in gene editing contexts.
In this experiment, the gRNA was designed to target the coding region of the mature HGF peptide. The editing event resulted in the insertion of six nucleotides, leading to the in-frame addition of two aspartic acid (DD) residues in the mature peptide. While this insertion does not disrupt the open reading frame, it likely compromises the structural integrity or functionality of the signal peptide, thereby impairing proper translocation and processing of the nascent HGF protein within the endoplasmic reticulum (ER) (Schröder and Kaufman, 2005). Consistent with this, our qRT-PCR and immunofluorescence analyses revealed a pronounced reduction in both HGF mRNA and protein levels in the knockout (KO-HGF) cells compared to controls, indicating successful disruption of HGF expression. To assess the functional impact of HGF depletion, we performed an MTS assay using ADSCs cultured with conditioned media from KO-HGF and negative control (NC-HGF) SCAPs. ADSCs exposed to KO-HGF conditioned media showed significantly reduced proliferation, demonstrating that the loss of HGF impairs the paracrine growth-promoting effect of SCAPs, in line with the established role of HGF in regulating cell proliferation. The observed downregulation of HGF mRNA and protein levels can be mechanistically attributed to the specific indel mutation introduced by CRISPR/Cas9 editing. Aberrant protein maturation within the ER is known to induce cellular stress responses, including activation of the unfolded protein response and ER-associated degradation pathways (Hetz, 2012; Sun and Brodsky, 2019). These quality control mechanisms serve to reduce the burden of misfolded proteins by downregulating gene expression at both transcriptional and post-transcriptional levels (Hetz et al., 2020; Miller et al., 2024; Ottens et al., 2024; Shin et al., 2024). Thus, the introduction of two negatively charged residues into the mature peptide likely disrupts normal HGF processing, triggering feedback mechanisms that compromise HGF mRNA stability and/or transcriptional activity (Hetz et al., 2020; Miller et al., 2024; Ottens et al., 2024; Shin et al., 2024). This provides a plausible explanation for the substantial reduction in both HGF mRNA and protein levels observed in the edited cells.
Conclusions
This study highlights ELNPs as a promising, efficient, and safe nonviral delivery platform for CRISPR/Cas9-mediated gene editing in SCAPs. Demonstrating high knockout efficiency with minimal cytotoxicity, ELNPs represent a viable alternative to conventional viral vectors and warrant further exploration for applications in regenerative medicine and gene therapy. Notably, ELNP-mediated delivery appeared to promote a unique or dominant DNA repair pattern following CRISPR/Cas9 editing, suggesting potential advantages for controlled genome modifications. However, the precise mechanisms driving this repair profile, as well as its broader applicability, require further investigation. Moreover, considering that EVs from different sources carry distinct RNA and circular DNA profiles, their use in gene editing may yield variable DNA repair outcomes. Therefore, systematic evaluation of EVs from diverse origins for each specific target gene is recommended to fully understand and optimize the potential of EV-based gene delivery in genome editing applications.
Authors’ Contributions
Conceived the experiments: S.E. and F.K. Designed the experiments: M.H.N.E., S.E., and F.K. Conducted the experiments: R.Y., S.E., and F.K. Provided resources: M.H.N.E., S.E., and F.K. Wrote the article: M.H.N.E., S.E., and F.K. Edited the article: M.N., S.E., and F.K.
Footnotes
Acknowledgments
The authors would like to appreciate all the staff of Royan Institute for Biotechnology.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This project was not supported by any national or international funding. No funding source was involved in the design or conduct of the research or preparation of the article, and the analyses and opinions expressed are those of the authors alone.
Ethics Declaration
The Research Committee of Royan Institute, Tehran, approved this project on April 3, 2022 (Certificate Number: IR.ACER.ROYAN.REC.1401.004). The committee was chaired by Dr. Abdolhossein Shahverdi, with Dr. Reza Omani Samani serving as the committee secretary.
Author Confirmation Statement
Each author confirms that this research is supported by an institution primarily dedicated to education or research.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.