
Abstract
Schlafen family member 11 (SLFN11), implicated in cancer drug resistance, may improve poly(ADP-ribose) polymerase inhibitor (PARPi) efficacy. This study investigates SLFN11’s role in epithelial ovarian cancer (EOC) progression and its influence on PARPi sensitivity, particularly in BRCA-wild-type contexts, with a focus on its emerging function in proteostasis regulation. SLFN11 expression in EOC and adjacent tissues was evaluated via immunohistochemistry, quantitative polymerase chain reaction (PCR), and Western blot. Functional assays—including cell viability, transwell migration, wound healing, and colony formation—assessed SLFN11’s effects on SKOV3 cells (BRCA-wild-type) proliferation and invasiveness. PARPi sensitivity in SKOV3 cells with SLFN11 knockdown or overexpression (OE) was tested using Cell Counting Kit (CCK)-8 and Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) assays. Protein stability and ubiquitination levels of PARP1/2 were analyzed as the central mechanism. SLFN11 mRNA and protein levels were markedly lower in EOC tissues compared with normal tissues. Silencing SLFN11 enhanced EOC cell proliferation, migration, and invasion, whereas OE inhibited these malignant behaviors. SLFN11 knockdown reduced PARPi-induced apoptosis and drug sensitivity, while its OE amplified these responses. Mechanistically, we demonstrate that SLFN11 suppresses global proteotoxic ubiquitination, thereby specifically stabilizing PARP1/2 proteins and potentiating PARPi-mediated DNA damage through enhanced chromatin trapping. SLFN11 enhances PARPi sensitivity in EOC by stabilizing PARP1/2 via inhibition of proteotoxic ubiquitination, supporting its role as a biomarker in a BRCA-wild-type EOC cell model where PARPi efficacy is limited by intrinsic resistance. While the application of PARPi in other subtypes of EOC still requires further validation, SLFN11 may improve PARPi response and could be explored as a strategy to address PARPi resistance.
Background
Ovarian cancer (OC) is the third most common and deadliest malignant tumor of the female reproductive system, with epithelial OC (EOC) accounting for >90% of cases. Approximately 70% of patients with OC are diagnosed at advanced stages (International Federation of Gynecology and Obstetrics [FIGO] stage III or IV), often characterized by extensive peritoneal dissemination and, in stage IV disease, distant metastases (Cortez et al., 2018). EOC comprises distinct histological subtypes—including high-grade serous ovarian carcinoma (HGSOC, 70%), endometrioid, clear cell, and mucinous carcinomas—each exhibiting unique molecular profiles and therapeutic responses. Despite standard treatment, which includes optimal cytoreduction followed by adjuvant chemotherapy, most patients experience recurrence and develop chemotherapy resistance, resulting in a global 5-year survival rate of ∼30–40% (Wang et al., 2024). Poly(ADP-ribose) (PAR) polymerase inhibitors (PARPi) have transformed treatment for BRCA-mutant or homologous recombination (HR)-deficient (HRD) HGSOC, significantly improving progression-free survival (González-Martín et al., 2023; Ray-Coquard et al., 2023). However, PARPi efficacy is limited in non-HGSOC subtypes (e.g., clear cell, mucinous) due to intrinsic resistance, while acquired resistance remains pervasive across all EOC subtypes (Cheng et al., 2022; Kulkarni et al., 2025). Intrinsic or acquired resistance to PARPi poses a significant challenge in the treatment of EOC and contributes to poor prognosis alongside other factors, such as high recurrence rates and resistance to chemotherapy.
PARPis have emerged as a promising targeted therapeutic strategy for EOC. To date, PARPi has been approved for the treatment of certain specific subtypes of EOC, primarily BRCA gene mutation carriers and homologous recombination-deficient (HRD)-positive high-grade serous carcinomas (Dal Molin et al., 2018; Moore et al., 2018). PARPi inhibits single-strand break repair by trapping PARP on DNA, thereby preventing PAR strand formation and subsequent DNA damage repair (DDR). This leads to replication fork stalling, accumulation of double-strand breaks, and failure of HR in DDR-deficient cells, ultimately causing mitotic catastrophe and apoptosis (Dziadkowiec et al., 2016; Sonnenblick et al., 2015). Clinical trials support the use of PARPi (e.g., olaparib, niraparib, rucaparib) as second-line or subsequent maintenance therapy for patients with platinum-sensitive recurrent OC (Mullen et al., 2019). While PARPi represents a significant advancement and is likely to be used by most patients, acquired resistance has emerged as a major clinical obstacle. Differentiating OC subtypes to expand PARPi efficacy and targeting efficient therapies are crucial for precision medicine. PARPi-resistant cell lines typically exhibit higher DDR capacity (Yamamoto et al., 2019), suggesting that increased DNA repair ability may contribute to resistance. Therefore, identifying new markers for HR repair deficiencies in EOC could help predict PARPi efficacy and extend its benefits to a broader patient population, ultimately improving survival outcomes.
Schlafen family member 11 (SLFN11), exclusively expressed in mammals, functions as a DNA and RNA deconjugating enzyme and has garnered increasing attention for its potential role in modulating the efficacy of anticancer drugs (Tang et al., 2018). When single-strand breaks occur in cellular DNA, SLFN11 protein binds to the DNA break site through replication protein A. Subsequently, its deconjugating enzyme alters the chromosome structure, causing the DNA replication fork to remain at the DNA break site, thus preventing DNA replication, leading to the failure of single-stranded DNA break repair (Murai et al., 2018), and, at the same time, inhibiting HR function for repairing cellular DNA damage (Mu et al., 2016). These findings suggest that SLFN11 may act as a synergistic lethal target for PARPi, potentially enhancing their antitumor effects. For example, in Lok et al. (2017), investigators found that small-cell lung cancer with high expression of the SLFN11 gene was sensitive to all three PARPis: olaparib, rucaparib, and veliparib. On the contrary, SLFN11-silenced cells were resistant to PARPi, and these were also verified using a tumor mouse model that the protein expression of SLFN11 and PARPi antitumor effects were positively correlated (Lok et al., 2017). In a retrospective analysis of 110°C patients, it was demonstrated that SLFN11 gene expression correlated with the efficacy of platinum-containing chemotherapy and that patients with high expression of the SLFN11 gene had a more pronounced overall survival benefit than those with low expression (Zoppoli et al., 2012). Given the correlation between SLFN11 methylation status and platinum chemotherapy outcomes, we propose that SLFN11 expression levels might similarly influence the efficacy of PARPis. Notably, recent studies have revealed that SLFN11 functions as a broader guardian of nuclear proteostasis, whose loss triggers global protein ubiquitination and endoplasmic reticulum stress (Iimori et al., 2025; Murai et al., 2021). This suggests that SLFN11 may modulate drug sensitivity by maintaining a cellular environment conducive to genome stability. Accordingly, we hypothesize that SLFN11 could enhance PARPi response in EOC by stabilizing key DNA repair proteins within this proteostatic framework. This study aims to validate SLFN11 as a predictive biomarker and elucidate its mechanistic role in PARPi sensitivity.
Accordingly, we hypothesize that SLFN11 expression could play a crucial synergistic role with PARPi in treating OC. Given its established correlation with platinum chemotherapy sensitivity, SLFN11 may also enhance PARPi response, particularly in tumors with HR deficiencies. This study aims to validate SLFN11 as a predictive biomarker, potentially extending the benefits of PARPi to a broader cohort of OC patients. Our study not only investigates the hypothesis that SLFN11 enhances PARPi sensitivity by stabilizing PARP1/2 but also explores the functional implications of this interaction on the DNA damage response and tumor cell survival. Despite prior studies linking SLFN11 expression to platinum chemotherapy sensitivity, its role in enhancing PARPi response remains underexplored (Onji et al., 2024). This research will address this gap, potentially transforming how we select OC patients for PARPi therapy and improving personalized treatment strategies.
Materials and Methods
Bioinformatics analysis of SLFN11-related data
Differences in SLFN11 gene expression between OC patients and healthy individuals were analyzed using the Gene Expression Profiling Interactive Analysis (GEPIA) database (http://gepia.cancer-pku.cn/). A boxplot illustrating SLFN11 gene expression levels was generated. Subsequently, the ENCORI database (https://starbase.sysu.edu.cn/) was utilized to examine the correlation in gene expression between SLFN11 and PARP1/2/3 in OC patients.
Clinical sample collection
The tissue samples analyzed in this study include 100 cases of OC tissue (64 serous carcinomas, 11 mucinous carcinomas, 4 endometrioid carcinomas, 17 clear cell carcinomas, 2 squamous cell carcinomas, 1 poorly differentiated adenocarcinoma, and 1 moderately differentiated adenocarcinoma), all obtained from patients with OC admitted to the Affiliated People’s Hospital of Ningbo University (Zhejiang, China) between December 1, 2016, and December 31, 2022. The protocol was reviewed and ethically approved by the Medical Ethics Committee of the Affiliated People’s Hospital of Ningbo University [Approval Number: 2021-(Research)-074], and all patients signed informed consent forms. The procedures and documentation of this study adhere to international ethical guidelines such as the International Ethical Guidelines for Health-Related Research Involving Humans and the Declaration of Helsinki, as well as relevant domestic laws and regulations.
Validation of SLFN11 protein expression in OC patients
Immunohistochemical techniques were employed to assess SLFN11 protein expression levels in OC tissue. The tissue microarrays were dewaxed and treated with citrate buffer (pH 6.0) at 121°C for antigen retrieval. They were then incubated with an anti-SLFN11 antibody (A20809, ABclonal, China; 1:1000) at 4°C for 16 h. After the primary antibody incubation, the tissue microarrays were treated with a goat antirabbit IgG-Horseradish Peroxidase (HRP) secondary antibody at 23 ± 3°C for 1 h. Nuclei were stained with hematoxylin following DAB color development. Photomicrographs were captured under a light microscope, and quantitative statistical analysis of optical density values was conducted using Image-Pro Plus 6.0.
Cell culture
Human EOC cell line SKOV3 (BRCA-wild-type), known for its low sensitivity to PARPi, was purchased from Zhejiang Ruyao Biotechnology Co., Ltd. (Ningbo, China). Cells were passaged in McCoy’s 5A medium containing 15% fetal bovine serum. The cells were incubated at 37°C in 5% CO2.
Construction of SLFN11 overexpression and knockdown vectors
The complete coding sequence of the SLFN11 transcript (NM_001376007.1) was synthesized by a commercial service (Azenta Life Sciences, Suzhou, China) and cloned into the pCDH-CMV-MCS-CopGFP-puro vector for overexpression (OE) studies. ShRNA sequences targeting SLFN11 were designed for knockdown experiments using the Broad Institute’s shRNA design portal (https://portals.broadinstitute.org/gpp/public/). To rule out off-target effects, three distinct shRNA sequences (shRNA1–3) were designed. To ensure knockdown specificity and rule out potential off-target effects, three distinct shRNA sequences were designed. The most effective construct, shSLFN11-#1, was selected for all subsequent functional experiments. A scrambled shRNA (silencing of the negative control [sh-NC]) served as the NC. Three shRNA sequences, along with a scrambled control, were synthesized by Azenta Life Sciences and cloned into the pLKO-U6-puro lentiviral vector using AgeI and EcoRI restriction sites. The sequences are listed in Table 1.
shRNA Sequences Used for SLFN11 Knockdown

Lentiviral packaging and transduction
Lentiviruses were produced by cotransfecting 293T cells with the respective shRNA or OE plasmids, along with packaging plasmids Δ8.91 and pVSV-G, using a cationic lipid transfection method (X-tremeGENE HP DNA transfection reagent, Roche, Switzerland). The medium was collected 48 h post-transfection, filtered, and used to infect SKOV3 cells. After 24 h of infection, the medium was replaced with McCoy’s 5A medium containing 2 µg/mL puromycin to select cells with stable viral integration over 7–9 days. Successfully transduced cells were maintained in the puromycin-free medium for further experimental analyses. OE-SLFN11 and knockdown efficiency in SKOV3 cells were validated by quantitative real-time PCR (qRT-PCR) and immunoblotting to assess mRNA and protein levels. Three independent biological replicates were performed for all experiments, with each replicate conducted on separate days using freshly passaged cells and reagents.
Quantitative real-time PCR
Quantitative PCR (qPCR) was conducted using SYBR qPCR SuperMix Plus (E096-01B, Novoprotein, China) on a 7500 Fast Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc., USA). Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) served as the internal reference gene for normalization. Relative gene expression was calculated using the 2−ΔΔCt method. The primer sequences used are listed in Table 2.
Primer Sequences for qRT-PCR

Western blot
Cells from various treatment groups were lysed using Radioimmunoprecipitation Assay (buffer) (RIPA) buffer containing 20 μL/mL protease inhibitors. Proteins were separated by Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) and transferred to a Polyvinylidene Fluoride (PVDF) membrane (ISEQ00010, Millipore, Germany). The membrane was blocked with 3% Bovine Serum Albumin (BSA) for 2 h at room temperature and incubated overnight at 4°C with primary antibodies against SLFN11 (A20809; 1:1000), Bcl-2 (A19693; 1:1000), Bax (A19684; 1:1000), PARP1 (A19596; 1:1000), PARP2 (A5945; 1:1000), PARP3 (A7297; 1:1000), ubiquitin (Ub; A19686 1:1000), and GAPDH (A19056; 1:1000) from ABClonal Company (Wuhan, China). After incubation with IgG-HRP secondary antibody (ab6721, 1:2000) at room temperature for 1 h, chemiluminescence was detected using an ECL system. Imaging was performed with a ChemiDoc-It Imaging System, and band intensity was quantified using ImageJ software. GAPDH was used for internal reference. Three independent biological replicates were performed for all experiments, with each replicate conducted on separate days using freshly passaged cells and reagents.
Cellular fractionation and chromatin-bound protein extraction
Chromatin-bound proteins were isolated using a modified subcellular fractionation protocol (Nuclear and Cytoplasmic Protein Extraction Kit, P0028, Beyotime, China). SKOV3 cells with modulated SLFN11 expression were treated with 66.67 μM Olaparib for 48 h. Approximately 1 × 107 cells per sample were harvested, washed with ice-cold Phosphate-Buffered Saline (PBS), and processed sequentially to obtain cytoplasmic, nucleoplasmic, and chromatin-bound fractions. Briefly, the cytoplasmic fraction was extracted first, followed by nuclear lysis to separate the nucleoplasmic supernatant. The final insoluble pellet, containing chromatin, was solubilized in RIPA buffer (P0013, Beyotime, China) to release chromatin-bound proteins. Protein concentrations were determined by Bicinchoninic Acid(BCA) assay (P0012, Beyotime, China). Equal amounts of protein from each fraction were analyzed by Western blot (see Section Western blot). Histone H3 and GAPDH served as markers for chromatin-bound and cytoplasmic fractions, respectively, to confirm fractionation specificity.
Cell scratch assay
SKOV3 cells overexpressing and silencing SLFN11, as well as NCs, were seeded onto 6 cm dishes. Once the cells reached confluence, a single straight scratch was made through the monolayer using a 200 μL pipette tip. The cells were then observed and photographed under a phase-contrast microscope at the initial time point, denoted as 0 h. Subsequently, the cells were allowed to continue incubating in the incubator, and the migration of cells at the marked position was observed under the phase-contrast microscope at 0 and 48 h. The results were quantified for cell migration distance using Image-Pro Plus 6.0 software. Three independent biological replicates were performed for all experiments, with each replicate conducted on separate days using freshly passaged cells and reagents.
Invasion assay
Cells (2 × 104 cells/well) were seeded into the upper chamber of the Transwell chamber (with an 8-μm pore size, purchased from Corning Inc.). The upper chamber was coated with Matrigel to form a uniform gel layer. The lower chamber of the Transwell was filled with 600 μL of complete growth medium. After a 48 h incubation period, cells in the lower chamber were stained with 1% crystal violet. The invasive ability of the cells was assessed by counting the number of cells in three distinct fields of view. Three independent biological replicates were performed for all experiments, with each replicate conducted on separate days using freshly passaged cells and reagents.
CCK-8 assay for cell viability
Cells were seeded in triplicate at 6000 cells/well in 96-well plates and incubated for 48 h. To assess cell viability, 10 μL of CCK-8 reagent (Dojindo, Japan) was added to each well, and the plates were incubated at 37°C for 2 h before measuring absorbance at 450 nm (OD450 nm) using a microplate reader (CMax Plus, MD, USA). For cytotoxicity testing, cells were treated with various concentrations of olaparib or rucaparib (0, 2.47, 7.4, 22.22, 66.67, 200, and 600 μM) for an additional 48 h. The inhibition rate of cell viability (%) was calculated as: (1 − OD450 nm experimental/OD450 nm control) ×100%. GraphPad Prism 8.0 was used to analyze inhibition rates and calculate IC50 values, indicating SKOV3 cell sensitivity to the drugs. Three independent biological replicates were performed for all experiments, with each replicate conducted on separate days using freshly passaged cells and reagents.
TUNEL staining
Overexpressed or silenced SKOV3 cells were treated with 22.22 μM rucaparib or 66.67 μM olaparib for 48 h, fixed with 4% paraformaldehyde, and stained for DNA fragmentation using a TUNEL staining kit (G1501, Servicebio, China). The stained cells were quantitatively analyzed using IPP6.0 software. Three independent biological replicates were performed for all experiments, with each replicate conducted on separate days using freshly passaged cells and reagents.
Protein ubiquitination degradation assay
SKOV3 cells overexpressing or silencing SLFN11 were treated with a protein synthesis inhibitor, cycloheximide (CHX, 50 μM), and a proteasome inhibitor, MG132 (50 μM), at 0, 2, 4, and 8 h. After treatment, cells were lysed, and the proteins were collected by centrifugation at 4°C and 15,000 × g for 20 min. Western blotting assessed changes in PARP1 and PARP2 protein expression levels and their ubiquitination status. In addition, cells transfected with 1.5 μg of plasmid encoding Flag-PARP1 or Flag-PARP2 and Hemagglutinin (tag) (HA)-Ub were cultured in RPMI-1640 medium without Fetal Bovine Serum (FBS) for 24 h. After replacing the medium, cells were incubated for an additional 24 h and then treated with MG132 (10 μM) for 2 h before harvesting for immunoprecipitation (IP). Ubiquitinated proteins were pulled down using Flag-(Protein) Tag (TAG) (Proteintech, 51064-2-AP) antibody and detected via Western blotting using Flag-TAG and HA-TAG (Proteintech, 66008-4-Ig) antibodies. Three independent biological replicates were performed for all experiments, with each replicate conducted on separate days using freshly passaged cells and reagents.
Statistical analyses
Data from three independent biological replicates were collected and presented as mean ± standard deviation. Statistical analyses were performed using GraphPad Prism version 8. For two-group comparisons, an unpaired two-tailed Student’s t-test was used. For comparisons involving more than two groups, one-way analysis of variance was conducted, followed by Tukey’s post hoc test when significant differences were found. Gene expression correlation analysis was performed using the Pearson correlation coefficient. A p value of <0.05 was considered statistically significant.
Results
Expression analysis of SLFN11 in EOC
The analysis conducted on the GEPIA website revealed that the SLFN11 gene was significantly downregulated in EOC tissues compared with normal human ovarian tissues, with the difference being statistically significant (p < 0.05, Fig. 1A). In addition, tissue microarray immunohistochemistry tests indicated that SLFN11 protein expression was significantly higher in adjacent normal tissues than in cancerous tissues, with a statistically significant difference noted (p < 0.01, Fig. 1B, C).
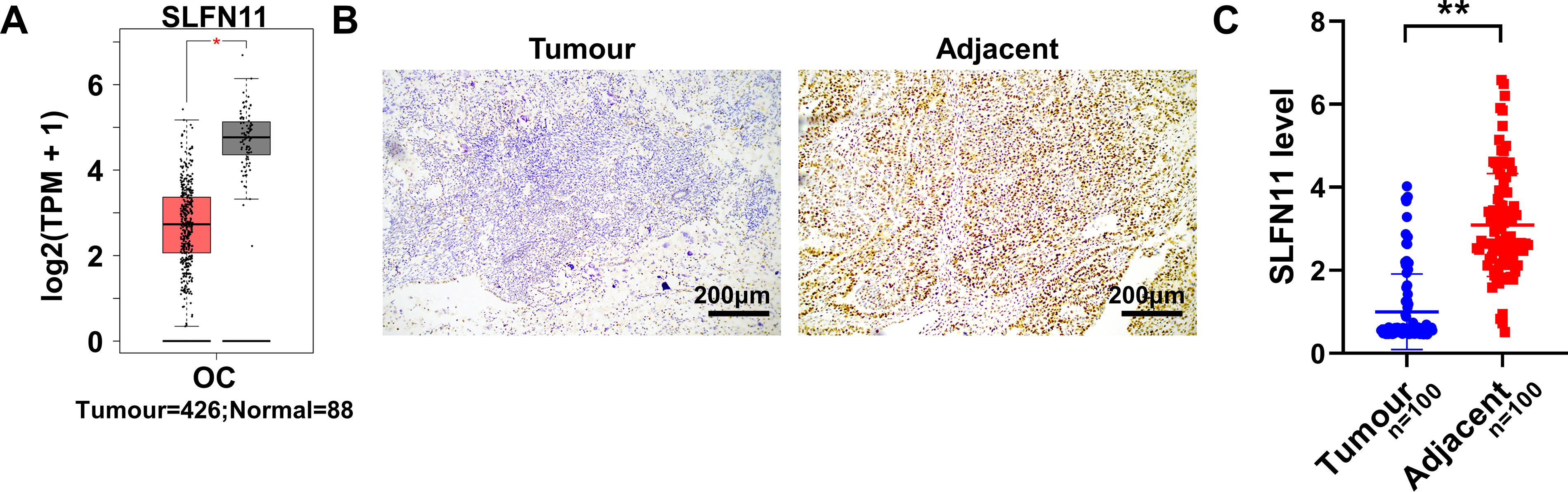
Analysis of SLFN11 expression levels in OC.
Effect of SLFN11 on the biological function of EOC cells
To explore the impact of SLFN11 on EOC progression, SKOV3 cells with engineered OE (OE-SLFN11) or silencing (sh-SLFN11) of SLFN11 were used. The results from qRT-PCR (Fig. 2A) and Western blot (Fig. 2B) demonstrated that OE-SLFN11 significantly increased the levels of the SLFN11 gene and protein to 2.5 ± 0.04-fold and 1.9 ± 0.16-fold, respectively. Conversely, sh-SLFN11-#1 reduced SLFN11 expression, with mRNA and protein levels reduced to ∼21% and 31% of the control, respectively. When compared with the NC group, these differences were statistically significant (p < 0.01). For all subsequent functional experiments (e.g., viability, migration, invasion, and drug sensitivity assays), the shSLFN11-#1 construct was used based on its robust knockdown efficiency. Following OE-SLFN11, cell viability (Fig. 2C), invasion rates (Fig. 2D, E), migration distances (Fig. 2F, G), and colony formation ability (Fig. 2H, I) were significantly decreased, all demonstrating statistically significant differences (p < 0.01) when compared with the OE-NC. In contrast, the silencing of SLFN11 led to a significant increase in cell viability, invasion rates, migration distances, and colony formation ability compared with the sh-NC, with each of these increases being statistically significant (p < 0.05). This indicates that even low SLFN11 expression constrains malignant behavior, and complete loss may exacerbate tumor aggressiveness.

Effects of SLFN11 on SKOV3 cellular functions.
SLFN11 regulates the susceptibility of SKOV3 cells to PARPi
CCK-8 assay results (Fig. 3A–D) indicate that OE-SLFN11 significantly reduced the IC50 values for rucaparib and olaparib from 33.21 and 298.8 to 18.81 and 103.2 μM, respectively. Conversely, silencing SLFN11 increased the IC50 values for these drugs from 33.62 and 305.1 to 60.84 and 690.4 μM, respectively. Rucaparib concentrations ranging from 7.40 to 66.67 μM actively inhibited the activity of SKOV3 cells overexpressing SLFN11 (p < 0.05 compared with the OE-NC group). In contrast, concentrations of 66.67–600 μM were less effective at inhibiting cell activity in cells with silenced SLFN11 (p < 0.01 compared with the sh-NC group). TUNEL staining (Fig. 3E–G) demonstrated that OE-SLFN11 significantly increased TUNEL positivity, indicating higher levels of DNA damage in SKOV3 cells treated with rucaparib and olaparib, with statistically significant differences compared with the NC+Ruca or NC+Ola groups (p < 0.01). Conversely, SLFN11 silencing significantly reduced the percentage of DNA damage in SKOV3 cells, showing a decrease in TUNEL positivity compared with the sh-NC group. However, the relationship between PARP1 levels and inhibitor efficacy, as suggested by our PARP1 OE data (Supplementary Fig. S1), is not linear and may vary according to the molecular context. It is plausible that the increased stability of PARP1/2 observed after OE-SLFN11 (see Fig. 4O-Q) may increase the amount of enzyme available to be “trapped” by PARPi, thereby enhancing cytotoxicity. This potential mechanism warrants further investigation to better understand how SLFN11 modulates PARPi sensitivity.
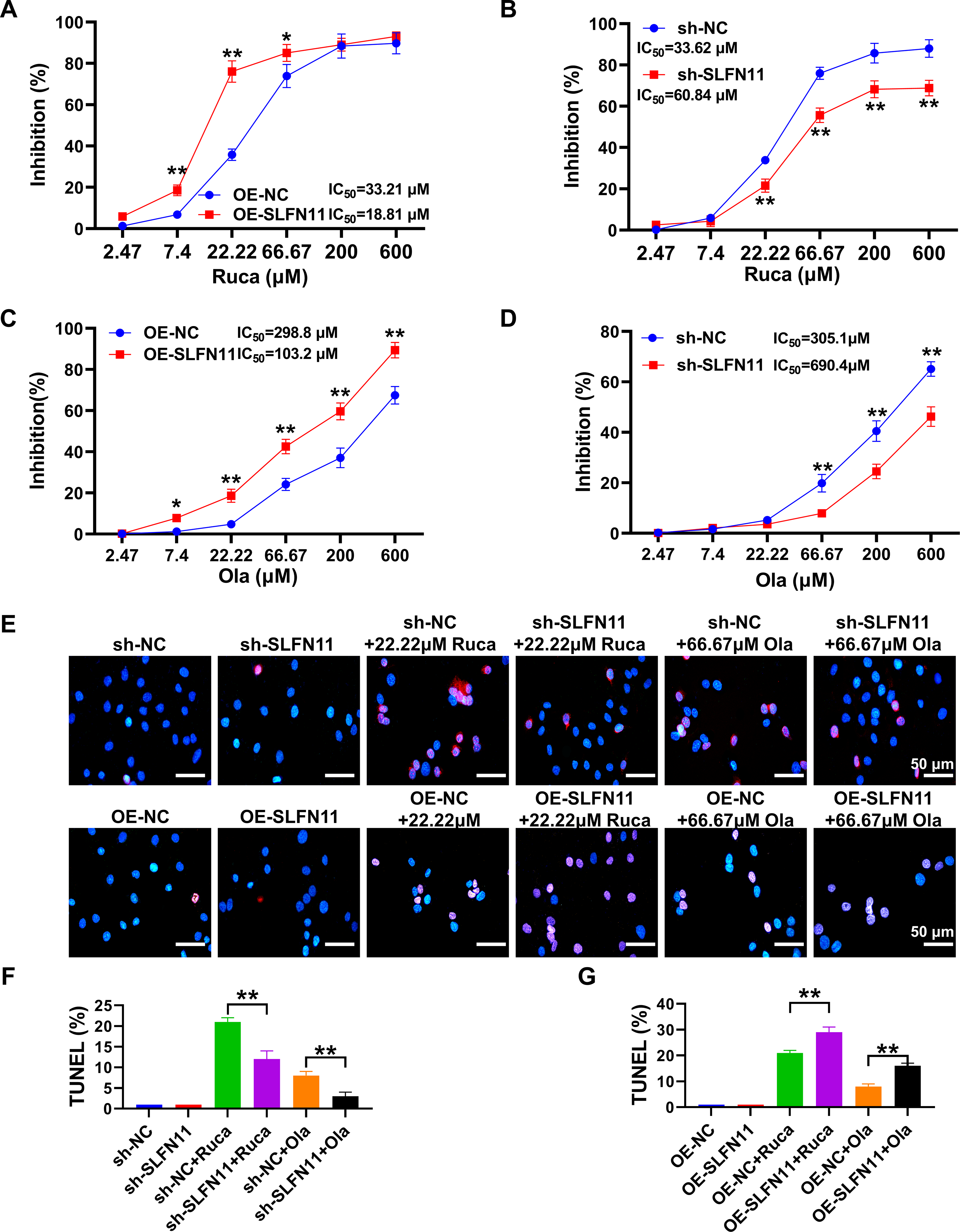
Study of SLFN11 differential expression levels on PARPi sensitivity.
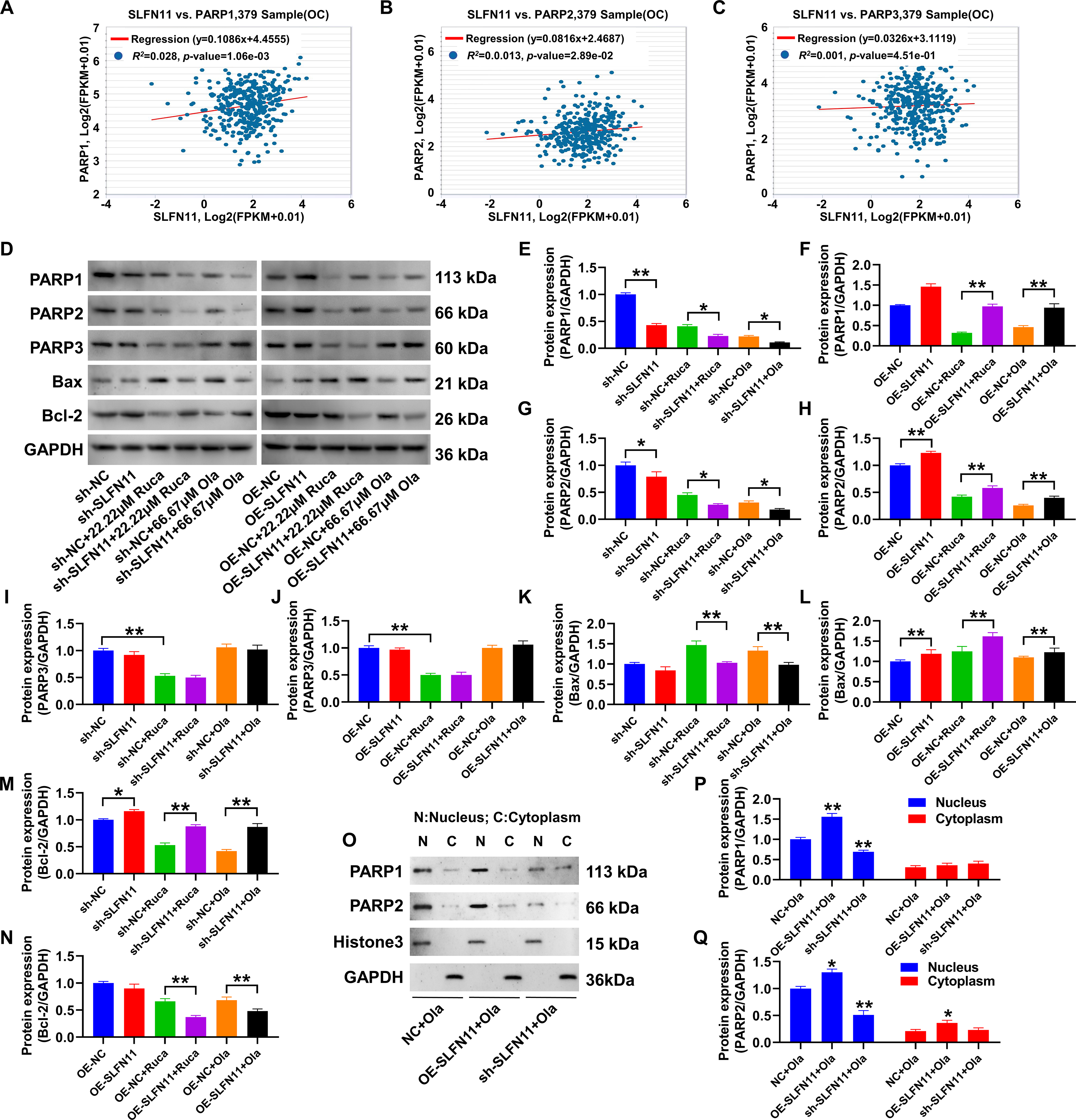
SLFN11 modulates expression of PARP isoforms and apoptosis-related proteins.
SLFN11 regulates PARP1/2 expression in EOC cells
Combining TCGA analysis and gene expression regulation data from the ENCORI database, SLFN11 showed a significant positive correlation with the expression levels of PARP1 and PARP2 genes (Fig. 4A-C). Western blot analysis (Fig. 4D-N) confirmed that overexpressing SLFN11 upregulated PARP1 and PARP2 expression in SKOV3 cells. Regardless of the presence of rucaparib or olaparib, elevated expression levels of PARP1/2 were observed compared with the OE-NC (p < 0.05). Silencing SLFN11 led to a marked reduction in PARP1/2 protein expression, significantly compared with the sh-NC (p < 0.05). However, SLFN11 did not influence PARP3 protein expression levels. Regarding apoptosis-related proteins, OE-SLFN11 resulted in a significant increase in the proapoptotic protein Bax, while the antiapoptotic protein Bcl-2 was significantly decreased, with statistically significant differences between the groups (p < 0.01). Conversely, silencing SLFN11 reduced Bax expression and increased Bcl-2 protein levels, again with statistically significant differences (p < 0.01). In addition, the Bax data showed statistical significance only when treated with the drugs, as indicated in Figure 4K (sh-NC vs. sh-SLFN11, not significant). In Figure 4N, the Bcl-2 protein levels in the OE-NC versus OE-SLFN11 group showed no statistically significant difference without drug treatment. OE-SLFN11 increased PARP1/2 stability, likely expanding the pool of PARP enzymes available for the “trapping process,” where PARPi locks PARP onto DNA lesions, converting repairable damage into lethal lesions. Critically, we provided direct functional evidence that elevating PARP1 or PARP2 levels is sufficient to sensitize cells to PARPi. OE of either PARP1 or PARP2 in SKOV3 cells significantly reduced the IC50 values for both rucaparib and olaparib. For rucaparib, the IC50 decreased from 28.59 μM in control cells (OE-NC) to 14.18 μM in OE-PARP1 cells and from 32.21 to 15.04 μM in OE-PARP2 cells (p < 0.05 at multiple concentrations, Supplementary Fig. S1B). For olaparib, the sensitization effect was even more pronounced, with the IC50 dropping from 311.3 μM in OE-NC cells to 44.19 μM in OE-PARP1 cells and from 305.1 to 51.87 μM in OE-PARP2 cells (p < 0.01 at multiple concentrations, Supplementary Fig. S1C). These results directly recapitulate the PARPi-hypersensitive phenotype observed in SLFN11-overexpressing cells, confirming that the stabilization of PARP1/2 is a key mechanistic step through which SLFN11 enhances PARPi efficacy. To further investigate whether the SLFN11-stabilized PARP1/2 proteins contribute to this canonical mechanism of PARP trapping, we performed cellular fractionation assays. SKOV3 cells with modulated SLFN11 expression were treated with olaparib, and the distribution of PARP1/2 in the chromatin-bound fraction was analyzed. As shown in Figure 4O, olaparib treatment induced a pronounced accumulation of PARP1 in the chromatin fraction of SLFN11-overexpressing cells (OE-SLFN11), which was significantly higher than that in the control cells (OE-NC) (Fig. 4P). Conversely, SLFN11 knockdown (sh-SLFN11) markedly attenuated the chromatin retention of PARP1 following olaparib exposure (Fig. 4O, P). A similar regulatory trend was observed for PARP2 (Fig. 4Q). These results demonstrate that SLFN11 expression level is a key determinant of the extent of PARP1/2 trapping on chromatin upon PARPi treatment, providing a direct mechanistic link between SLFN11-mediated PARP1/2 stabilization and enhanced PARPi cytotoxicity. These findings indicate that SLFN11 plays a crucial role in regulating the stability and function of PARP1/2, thereby influencing the response to PARPi.
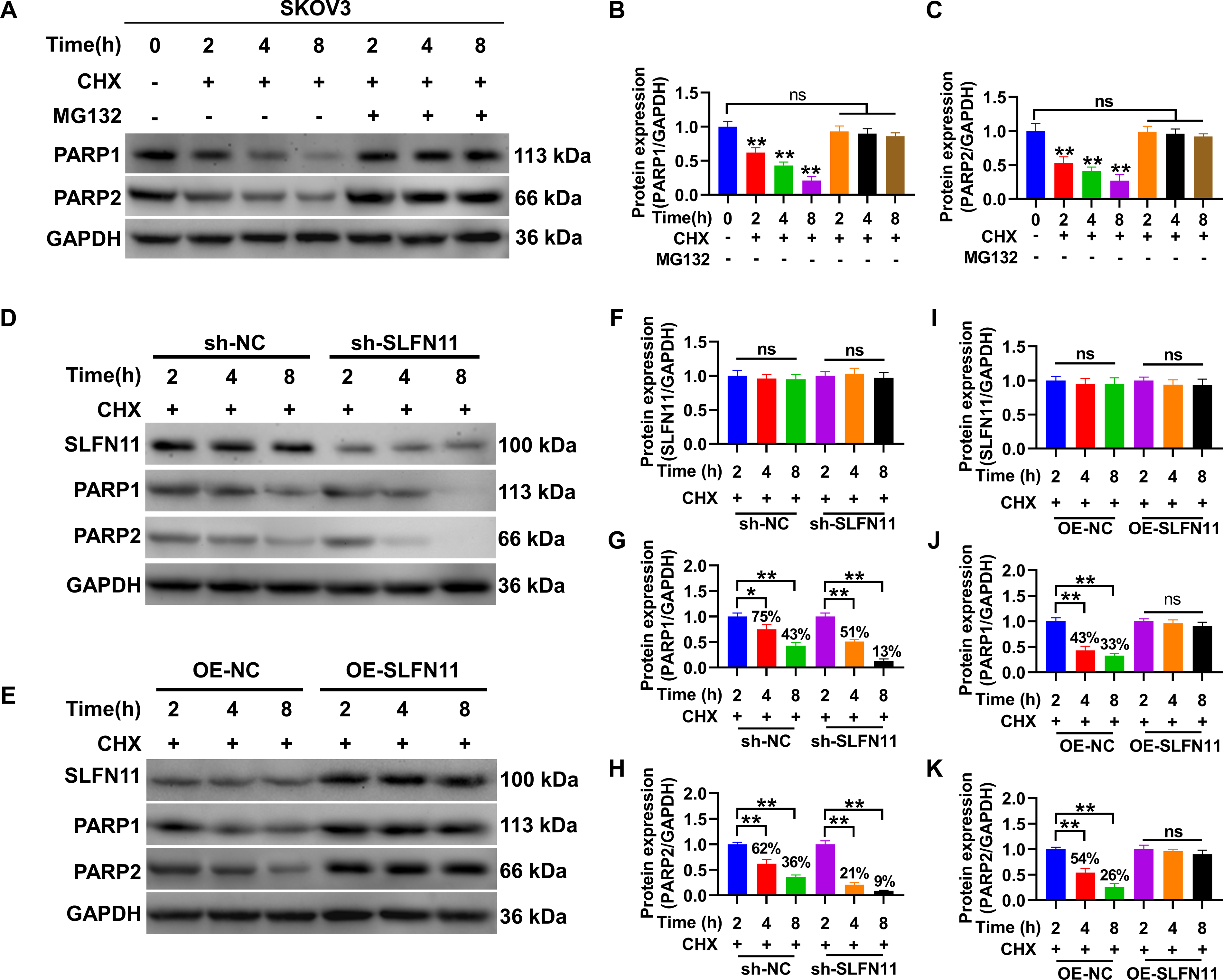
The impact of SLFN11 on the degradation efficiency of PARP1 and PARP2 proteins.
SLFN11 regulates the degradation rate of PARP1/2 proteins
the differences were statistically significant compared with the 0-h time point (p < 0.01). The addition of the proteasome inhibitor MG132 effectively prevented this change (Fig. 5A–C). Furthermore, by constructing SLFN11 knockdown and OE cells, the effect of SLFN11 expression on the degradation rate of PARP1/2 proteins was analyzed. Immunoblotting results showed that in SKOV3 cells with silenced SLFN11 compared with the sh-NC group, the content of PARP1 and PARP2 proteins decreased from 43% and 36% to 13% and 9% within 8 h, respectively (Fig. 5D–I), indicating a significantly increased degradation rate. In addition, in the OE-NC group, the content of PARP1 and PARP2 proteins decreased to 33% and 26%, respectively, while in cells overexpressing SLFN11, the content of PARP1 and PARP2 proteins showed no significant change after 2 h (p > 0.05) (Fig. 5E–K), indicating a significantly reduced degradation rate. These results suggest that SLFN11 can regulate the stability of PARP1/2 proteins through the proteasome system.
SLFN11 regulates the ubiquitination levels of PARP1/2 proteins
When investigating the mechanism by which SLFN11 affects the stability of PARP1/2 proteins, SKOV3 cells were co-transfected with Flag-PARP1/2 and HA-Ub plasmids, and anti-Flag antibody was used for IP to assess the impact of OE-SLFN11 or silencing on the ubiquitination levels of PARP1/2. The results of the immunoblotting analysis showed that under the co-expression of HA-Ub, the OE-SLFN11 led to a significant decrease in the ubiquitination levels of Flag-PARP1/2 (p < 0.05), as shown in Figure 6A–D. In cells where SLFN11 was silenced, the ubiquitination levels of Flag-PARP1/2 significantly increased when co-expressed with HA-Ub (p < 0.01), as shown in Figure 6E-H. These results indicate that SLFN11 plays a key role in regulating the ubiquitination levels of PARP1/2, and the silencing of SLFN11 promotes the ubiquitination process of PARP1/2. While our study did not identify the specific effector molecules downstream of SLFN11, the observed reduction in global ubiquitination upon OE-SLFN11 strongly suggests that it functions as a master regulator of the Ub–proteasome system. We postulate that SLFN11 may achieve this by either suppressing the activity of specific E3 Ub ligases that target PARP1/2 for degradation or by enhancing the function of deubiquitinating enzymes that stabilize them. This selective inhibition of PARP1/2 ubiquitination represents a precise molecular strategy to increase the cellular pool of these proteins, thereby amplifying the PARP-trapping effect and enhancing PARPi sensitivity.
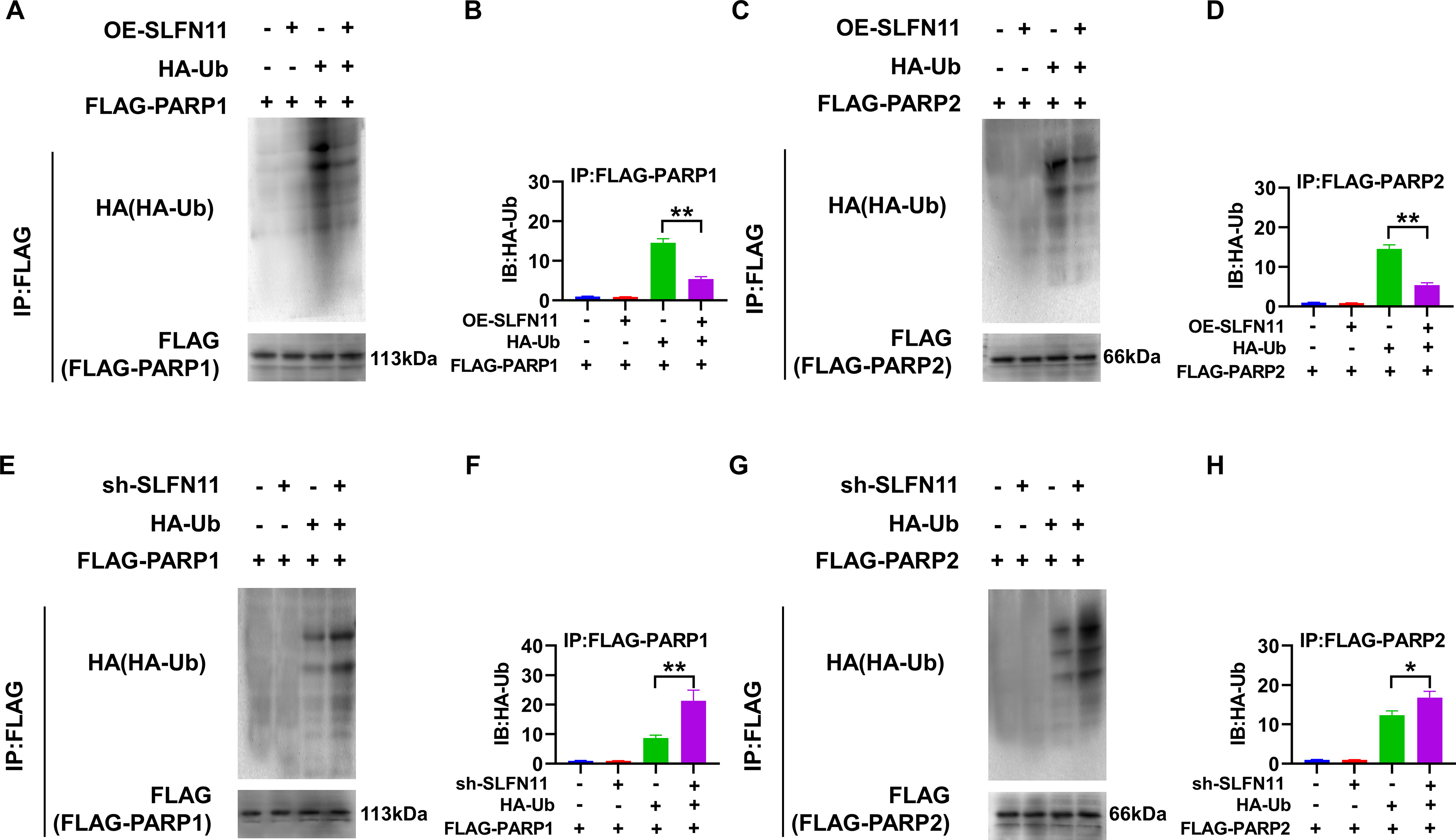
Regulation of PARP1 and PARP2 Ubiquitination Levels by SLFN11.
SLFN11 modulates the global ubiquitination landscape and differentially affects PARP family members
To investigate whether SLFN11’s effect on PARP1/2 ubiquitination is part of a broader regulatory function, we assessed its impact on global protein ubiquitination. SKOV3 cells with modulated SLFN11 expression were treated with the proteasome inhibitor MG132 to enrich for ubiquitinated substrates. As shown in Figure 7A, B, SLFN11 knockdown (sh-SLFN11) led to a significant accumulation of poly-ubiquitinated proteins compared with its control (sh-NC). Conversely, OE-SLFN11 resulted in a marked reduction of global ubiquitination relative to its control (OE-NC). Western blot analysis of lysates from cells with or without MG132 treatment consistently revealed the same trend. While the overall protein levels increased after MG132 treatment across all groups, the relative expression pattern remained consistent. This demonstrates that SLFN11 acts as a suppressor of aberrant, degradation-prone ubiquitination in EOC cells.
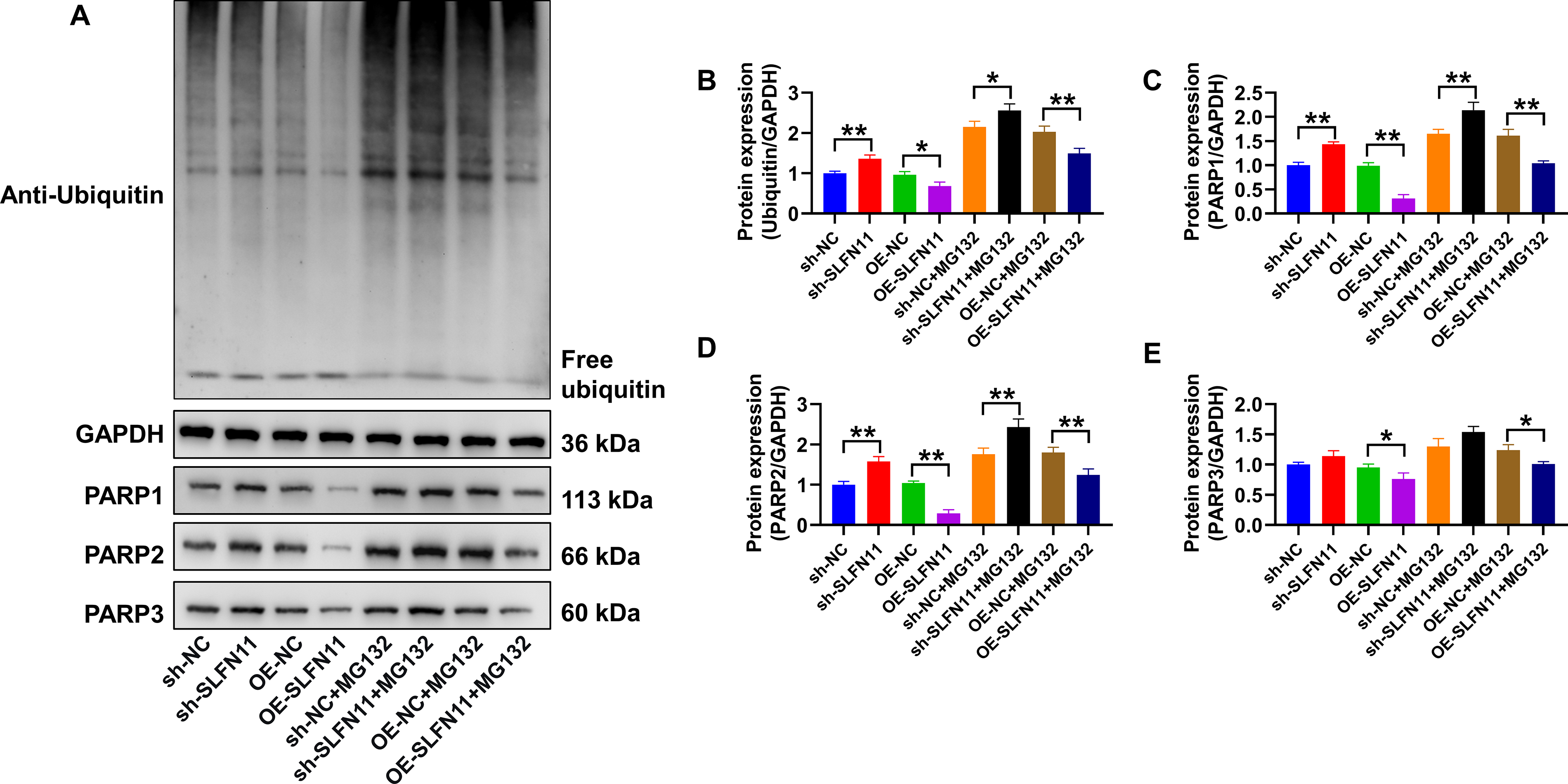
SLFN11 modulates global ubiquitination and differentially regulates PARP family protein stability.
We then examined how this global change in the ubiquitination environment specifically influenced different PARP family members. Quantification confirmed that the protein levels of PARP1 and PARP2 strongly correlated with SLFN11 expression, decreasing upon knockdown and increasing upon OE (Fig. 7C, D). This regulatory pattern was highly significant (p < 0.01) between all corresponding control and SLFN11-modulated groups, both with and without MG132, and is consistent with their high sensitivity to the ubiquitination landscape. In stark contrast, the abundance of PARP3 remained essentially unchanged across most conditions, displaying only subtle variations, with statistical significance detected in only two-group comparisons (p < 0.05) (Fig. 7E). These results indicate that PARP1 and PARP2 are particularly susceptible to the SLFN11-modulated proteostatic environment, whereas PARP3 appears to be relatively resilient. The findings suggest that PARP1 and PARP2 are particularly vulnerable to Ub-mediated degradation, and their stability is highly dependent on the protective, anti-ubiquitination function exerted by SLFN11.
Validation of the SLFN11-PARP1/2 regulatory axis in a second EOC model
To ensure the generalizability of our findings, we validated the core observations in OVCAR3, another well-characterized BRCA-wild-type EOC cell line. We established OVCAR3 cells with stable OE-SLFN11 or knockdown. Consistent with the results in SKOV3 cells, TUNEL assays revealed that OE-SLFN11 significantly enhanced PARPi-induced apoptosis, while SLFN11 knockdown conferred resistance (Supplementary Fig. S2A–C). Furthermore, Western blot analysis confirmed that the regulatory relationship between SLFN11 and its downstream targets was conserved in OVCAR3 cells. SLFN11 expression positively correlated with the protein levels of PARP1, PARP2, and the proapoptotic protein Bax and negatively correlated with the antiapoptotic protein Bcl-2, particularly upon PARPi treatment (Supplementary Fig. S2D–H). These concordant results across two distinct EOC models strongly suggest that the role of SLFN11 in modulating the PARP1/2-Bax/Bcl-2 axis to potentiate PARPi sensitivity represents a broader mechanism in EOC, rather than a cell line-specific phenomenon.
Discussion
Sensitivity to PARPi in EOC models depends on SLFN11 expression, with high levels correlating with a positive response. In contrast, resistance is frequently observed when SLFN11 expression is low or absent (Jo et al., 2021; Zhang et al., 2021). Despite these associations, the mechanistic link between SLFN11 expression and PARPi sensitivity in EOC remains largely unexplored. Furthermore, clinical observations, such as varying responses to PARPi among patients with similar SLFN11 levels, have yet to be fully explained (Willis et al., 2021). This gap underscores the significance of our study, which aims to provide insights into therapeutic strategies for OC patients with varying levels of SLFN11 expression and to elucidate its mechanistic role in PARPi sensitivity and chemoresistance. PARPis can selectively target BRCA-mutated EOC, although they also have some effect on some EOC cell lines without BRCA mutations. For instance, the A2780 cell line, which lacks BRCA mutations, can still have its proliferative activity and invasiveness significantly inhibited by PARPis olaparib or AG14361 (Xiang et al., 2021). However, in Xiang et al.’s study, the inhibitory effect of PARPis on the proliferative activity of SKOV3 cells, which are also BRCA wild-type, is almost negligible (Xiang et al., 2021). The OVCAR3 cell line, although exhibiting resistance to olaparib (Lin et al., 2018a), shows a higher drug sensitivity to another PARPi, rucaparib (Bradbury et al., 2020). Many studies have also focused on the PARPi resistance characteristics of the SKOV3 cell line, including but not limited to rucaparib and olaparib (Ma et al., 2022a, b). The SKOV3 cell line demonstrates a more tenacious resistance to PARPis. Therefore, using the SKOV3 cell line is of significant importance for elucidating the PARPi resistance mechanisms in some OC patients and is an important research cell line for PARPi resistance in EOC.
In our analysis, silencing and OE-SLFN11 in SKOV3 cells markedly influenced cellular behaviors: OE was associated with significantly reduced proliferation, migration, and invasion, akin to observations in liver cancer (Zhou et al., 2020). SLFN11 primarily regulates DNA damage induction, stalling replication forks, and activating cell cycle checkpoints—a phenomenon termed replication stress. Recent studies highlight that SLFN11 is recruited to replication forks under such stress, effectively halting replication and enhancing the cytotoxicity of replication stress-inducing drugs (Murai et al., 2018, 2019). PARPi, known for its significant survival benefits in various tumors, including EOC, functions by inhibiting the repair of single-strand breaks, potentially enhancing apoptosis in tumor cells (Cortez et al., 2018). Our findings reveal that OE-SLFN11 augments EOC cell sensitivity to rucaparib and olaparib, with a concurrent increase in apoptosis levels. Thus, SLFN11 is a crucial marker for assessing PARPi response in EOC cells.
However, the specific mechanisms through which SLFN11 influences PARPi sensitivity remain to be clarified. It is widely believed that a complementary mechanistic action exists between SLFN11 and PARPs. Our study confirms this, showing a positive correlation between SLFN11 and PARP1/PARP2 proteins through Western blot analysis. We propose that SLFN11’s role in inducing DNA damage and the subsequent cellular adaptation involves the upregulation of PARPs to enhance cell viability under replication stress. Nevertheless, the intricate regulatory relationship between SLFN11 and PARPs warrants further investigation to fully elucidate their coordinated role in modulating PARPi efficacy.
The cellular protein levels are determined by the balance between synthesis and degradation rates. Protein half-lives can vary significantly, ranging from minutes to days, reflecting their diverse roles in cellular metabolism. Rapidly degraded proteins, such as transcription factors, are essential for quick cellular responses to external stimuli due to their regulatory functions. In addition, specific signals can trigger the rapid degradation of other proteins, providing a mechanism to regulate intracellular enzyme activities. The primary pathways for protein degradation include the Ub–proteasome system and lysosomal protein hydrolysis (Munro et al., 2024). In the study by Murai et al. (2021), SLFN11 knockdown in various cancer cell lines, including DU145 (prostate cancer) and DMS114 (small-cell lung cancer), resulted in a significant increase in global ubiquitination levels. This finding suggests that SLFN11 regulates ubiquitination processes, potentially influencing protein stability and degradation pathways. Elevated global ubiquitination levels observed in SLFN11-deficient cells may lead to increased protein aggregation, contributing to heightened vulnerability to proteotoxic stress. This observation highlights SLFN11’s protective role in mitigating the accumulation of ubiquitinated proteins. Moreover, SLFN11 deficiency has been linked to increased sensitivity to proteotoxic stress, further underscoring its role in maintaining cellular homeostasis under stress conditions. These characteristics suggest that SLFN11 may serve as a potential target for modulating PARPi sensitivity in cancer therapy, particularly in EOC and other SLFN11-deficient tumors. By understanding the regulatory role of SLFN11 in ubiquitination and its impact on drug resistance, we can better delineate its potential in advancing therapeutic strategies.
To validate this hypothesis, we employed the proteasome inhibitor MG132 and protein synthesis inhibitor CHX. Ubiquitination is a key pathway for protein degradation, and MG132 inhibits the proteasome-dependent degradation of ubiquitinated proteins by targeting the β5 catalytic subunit of the 20S proteasome, leading to the accumulation of ubiquitinated substrates (Sun et al., 2018). CHX, on the other hand, suppresses ribosomal translation, halting de novo protein synthesis. This suppression prevents newly synthesized, nonubiquitinated proteins from diluting pre-existing ubiquitinated proteins, thereby allowing us to observe their accumulation. Simultaneous inhibition of protein synthesis and proteasome activity enables the study of the balance between protein synthesis and degradation (Miao et al., 2023). MG132 and CHX are frequently used in EOC research to investigate ubiquitination pathways and their role in regulating protein stability (Ali et al., 2019; Lin et al., 2018b; Zhu et al., 2023). Our results demonstrate that OE-SLFN11 reduces the degradation and ubiquitination of PARP1 and PARP2 proteins, thereby stabilizing these proteins and confirming SLFN11’s role in regulating their proteasome-dependent degradation.
Our study demonstrates that SLFN11 enhances PARPi sensitivity by specifically inhibiting the ubiquitination and degradation of PARP1/2. This finding gains profound mechanistic context when viewed through the lens of SLFN11’s newly recognized function as a global regulator of proteostasis (Murai et al., 2021). We found that SLFN11 knockdown elevates global protein ubiquitination, while its OE suppresses it, confirming its role as a buffer against proteotoxic stress in EOC.
Combining our findings, we propose a coherent model: SLFN11 inhibits the Ub-mediated degradation of PARP1 and PARP2, thereby increasing their cellular abundance. Crucially, as directly evidenced by our chromatin fractionation data, this enlarged pool of PARP proteins leads to a more profound “trapping” of PARP1/2 on chromatin upon PARPi treatment. This synergistic action results in the stabilization of cytotoxic DNA–protein complexes, accumulation of irreparable DNA damage, and, ultimately, enhanced apoptosis in cancer cells (Kim et al., 2020; Murai et al., 2012). Critically, we provide direct functional evidence for this model by demonstrating that artificial OE of PARP1 alone is sufficient to recapitulate the PARPi-hypersensitive phenotype. These complementary lines of evidence establish a transparent causal chain from SLFN11 expression to PARP1/2 stabilization and finally to enhanced PARPi cytotoxicity via augmented PARP trapping.
SLFN11 stabilizes PARP1/2 within a broader role as a guardian of proteostasis. The mechanism by which SLFN11 regulates PARP1/2 ubiquitination can be understood within the emerging paradigm that positions SLFN11 as a master regulator of nuclear and cellular proteostasis (Murai et al., 2021). Seminal studies have shown that SLFN11 inactivation leads to increased global protein ubiquitination, ER stress, and heightened sensitivity to proteotoxic stressors, establishing its role in mitigating protein quality control crises (Iimori et al., 2025; Murai et al., 2021). Furthermore, SLFN11 induces cell death under DNA damage by impairing translation via ribosome stalling and tRNA cleavage, pathways that inherently disrupt proteostasis (Boon et al., 2024; Ogawa et al., 2025). In this light, our discovery that SLFN11 specifically suppresses the ubiquitination and degradation of PARP1/2 likely represents a targeted manifestation of this broader, protective function (Xiao et al., 2025).
We therefore propose a unified model: SLFN11 maintains a cellular environment resistant to aberrant ubiquitination. Within this protected milieu, functionally critical yet metabolically vulnerable proteins are stabilized. PARP1 and PARP2, due to their high nuclear abundance, constant enzymatic activity, and essential role in DNA repair, represent prime examples of such proteins (Lin et al., 2025; Wang et al., 2012). Their stabilization leads to an enlarged enzyme pool available for PARPi-induced “trapping” on chromatin. The selectivity of this effect—sparing PARP3—likely stems from PARP3′s lower expression and distinct functional niche, rendering it less susceptible to the same proteostatic pressures (van Beek et al., 2021), as reflected in our data.
This model elegantly bridges SLFN11’s canonical role in DNA damage response with its emerging function in protein quality control. It explains why SLFN11 expression is such a potent biomarker: it directly reports on the cell’s capacity to maintain the stability of key therapeutic targets like PARP1/2 under genotoxic stress. Our work positions the SLFN11-PARP1/2 axis as a critical interface between proteostatic resilience and drug response, offering a new framework for understanding and overcoming PARPi resistance.
Conclusion
SLFN11 potentially inhibits EOC progression by stabilizing PARP1/2 protein levels through reduced ubiquitination. This stabilization enhances the efficacy of DNA-damaging agents, including platinum-based drugs and PARPi, by increasing apoptosis and suppressing tumor growth. However, our study primarily focused on the correlation between SLFN11 and PARP ubiquitination without identifying the specific Ub ligases or deubiquitinating enzymes regulated by SLFN11. To translate our findings into clinical practice, a critical next step is to identify the specific E3 ligases or deubiquitinating enzymes through which SLFN11 regulates PARP1/2 stability. Elucidating this precise molecular axis will not only deepen our understanding of proteostasis in therapy response but also reveal novel predictive biomarkers to better stratify patients. Furthermore, it may uncover new therapeutic targets whose inhibition could mimic the SLFN11-high state, thereby resensitizing SLFN11-low, PARPi-resistant tumors to therapy. We acknowledge several limitations in our study. The use of the SKOV3 cell line, while relevant for its BRCA-wild-type and PARPi-resistant characteristics, may limit the generalizability of our findings to other EOC subtypes. In addition, the absence of in vivo validation is a notable limitation, as it prevents a comprehensive assessment of the impact of SLFN11 on PARPi response in a more clinically representative context. The tumor microenvironment and pharmacodynamics, which play crucial roles in therapeutic efficacy, cannot be fully captured in in vitro models. Furthermore, our study lacks correlation with clinical data, which would strengthen the translational relevance of our findings. Future research should incorporate in vivo models and clinical cohorts to address these limitations and provide a more complete picture of SLFN11’s role in EOC.
Authors’ Contributions
M.L. designed the experimental scheme and served as the corresponding author, overseeing the submission process and communication with the journal. Material preparation, data collection, and analysis were performed by M.L., H.L., F.Z., and L.F. The first draft of the article was written by M.L. All authors commented on previous versions of the article. L.F. reviewed the data and manuscripts and was responsible for supervising the experiment. L.F. provided additional correspondence and oversight. All authors read and approved the final article.
Footnotes
Acknowledgments
The authors thank Zhejiang Ruyao Biotechnology Co., Ltd. (Ningbo, China).
Funding Information
This study was supported by the Zhejiang Provincial Medical and Health Science and Technology Plan Project (No. 2022KY1180), the Natural Science Foundation of Ningbo Municipality (No. 2024J380), the Agriculture and Social Development Project of Yinzhou District (No. 2024AS057), and the Ningbo Health Science and Technology Program (No. 2022y39).
Data Availability
The dataset generated during and/or analyzed during the current study is available from the corresponding author upon reasonable request.
Disclosure Statement
The authors have no conflicts of interest to declare.
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.