
Abstract
Background:
DUOX2 is a major cause of congenital hypothyroidism (CH) in Chinese patients, but clinical outcomes for those with biallelic DUOX2 mutations remain unclear. This study aimed to describe the clinical manifestations of CH due to DUOX2 defect.
Methods:
One hundred eighty-one patients with primary CH were recruited initially and were subjected to genetic screening. Patients with DUOX2 biallelic mutations were chosen. After 3 years of age, 28 patients underwent a prospective clinical reevaluation after levothyroxine (LT4) withdrawal. Subsequent periodic evaluation of thyroid function was executed to evaluate the necessity of LT4 retreatment. The medical histories of all patients before the age of three years were collected and analyzed. DUOX2 residual enzymatic activity was also calculated relative to clinical outcomes.
Results:
Twenty-eight patients who were reevaluated were divided into three groups: patients with permanent CH (PCH; 7/28 [25%]), patients with transient CH (TCH; 6/28 [21.4%]), and patients with hyperthyrotropinemia (15/28 [53.6%]). The median duration of follow-up was 17.5 months (interquartile range: 8.5, 29.25). The correlation between DUOX2 residual enzymatic activity and the clinical outcome of patients with CH with DUOX2 biallelic mutations was not clear in this study. No significant differences in laboratory findings at diagnosis were observed among the three groups. LT4 dose decreased with age in TCH but remained stable in PCH. Doses at ages 2, 3, and pre-withdrawal were significantly higher in PCH versus TCH (p = 0.027; p = 0.003; p = 0.025). After LT4 withdrawal, serum thyroglobulin levels and thyroid size increased in most patients (especially hyperthyrotropinemia group) and often persisted for months. Moreover, thyrotropin levels normalized in 44.4% of patients with hyperthyrotropinemia after more than one year off LT4.
Conclusions:
Some patients with CH and DUOX2 biallelic mutations may have TCH or hyperthyrotropinemia. These patients should undergo long-term follow-up to prevent excessive compensatory thyroid hyperplasia.
Introduction
Congenital hypothyroidism (CH), a common pediatric endocrine disease, causes intellectual disability and growth retardation if treatment is delayed. 1,2 Timely supplementation with appropriate doses of levothyroxine (LT4) prevents irreversible intellectual and linear growth retardation in patients with CH.
During treatment, parents of patients with CH are mostly concerned about the clinical outcomes of their children, namely, whether their children would need lifelong therapy and the correct time, if any, to withdraw therapy. In most studies, the clinical outcomes of CH have been classified as permanent CH (PCH) or transient CH (TCH). The current protocol for distinguishing between PCH and TCH is to reevaluate the patient’s thyroid function by LT4 withdrawal at approximately three years of age. 3 –5 Apart from these two forms of CH, recent studies have reported that some patients with CH present intermediate thyroid function, referred to as hyperthyrotropinemia, which is characterized by thyrotropin (TSH) levels above the upper limit of the reference interval but <10 µIU/mL and normal free thyroxine (fT4) levels. 6 –8 Given the management of persistent hyperthyrotropinemia was controversial, there was insufficient evidence to determine whether the treatment with LT4 was of clinical benefit. 4 Thus, in this study, the CH patients with slight increase in TSH (greater than the age-specific reference range, but < 10µIU/mL) and normal in fT4 during the LT4 trial off test have been suggested to continue withdrawal of LT4 therapy and retest the thyroid function in another 3–4 weeks in order to observe the clinical outcomes of these patients.
A consensus was reached that patients with PCH should remain on lifelong LT4 therapy, while those with TCH could discontinue therapy if they are aged older than three years. However, a recent study reported that LT4 therapy was reintroduced in two patients with TCH due to significant increase of TSH levels (17 mIU/L and 9.2 mIU/L, respectively) after withdrawal of LT4 therapy for 2 and 9 years. 6 Therefore, it was unclear whether patients with TCH could discontinue LT4 treatment lifelong. Moreover, it is unclear whether patients with CH and hyperthyrotropinemia require LT4 treatment after three years of age. 9
Many studies have reported that the clinical outcomes of CH are associated with genetic factors, such as DUOX2 mutation. 7,9 –11 Our previous studies have shown that DUOX2 is the most common causative gene of CH, with approximately 40% of patients with CH in China carrying DUOX2 biallelic mutations. 12 In this study, clinical reevaluation after T4 withdrawal and long-term follow-up were performed to describe the clinical evolution of CH in patients with DUOX2 biallelic mutations.
Methods
Patients
One hundred eighty-one patients with CH were identified through newborn screening utilizing a blood sample obtained from the baby’s heel between 48 and 72 hours of life. The blood spot TSH cutoff value was 10 µIU/mL. When TSH exceeded the threshold, a confirmatory test on neonatal serum to measure levels of TSH, T3, T4, fT3, and fT4 by an immunochemiluminometric assay (UniCel DxI 800, Beckman, USA) was done. Diagnosis of primary CH was confirmed by elevated serum TSH with or without lower fT4 levels according to laboratory reference values.
Genetic screening
After diagnosing with CH, patients were subjected to genetic screening for the causative genes of CH by targeted next-generation sequencing of 21-gene panel or whole exome sequencing as before. 12,13 Based on the sequencing results, patients with multiple DUOX2 variants or homozygous variants were selected, and family studies were performed to verify DUOX2 biallelic mutations.
Inclusion and exclusion criteria
Patients finally enrolled in this study must simultaneously meet the following inclusion criteria: i) diagnosis with primary CH; ii) DUOX2 gene as the causative gene after genetic screening; and iii) age more than three years old. The following CH patients should be excluded: i) patients with CH younger than three years of age; ii) patients with CH who also carried biallelic mutations of other CH-related genes except DUOX2; iii) patients with CH with central hypothyroidism and thyroid dysgenesis, including agenesis and ectopic and hypoplastic thyroid gland. Based on sequencing results, 61 patients carried DUOX2 biallelic mutations, of which 4 also combined biallelic mutations of other related genes that were excluded (Fig. 1). This study was approved by the Ethics Committee of Shanghai Ninth People’s Hospital affiliated with Shanghai Jiao Tong University School of Medicine (approval number: SH9H-2016–76-T33).
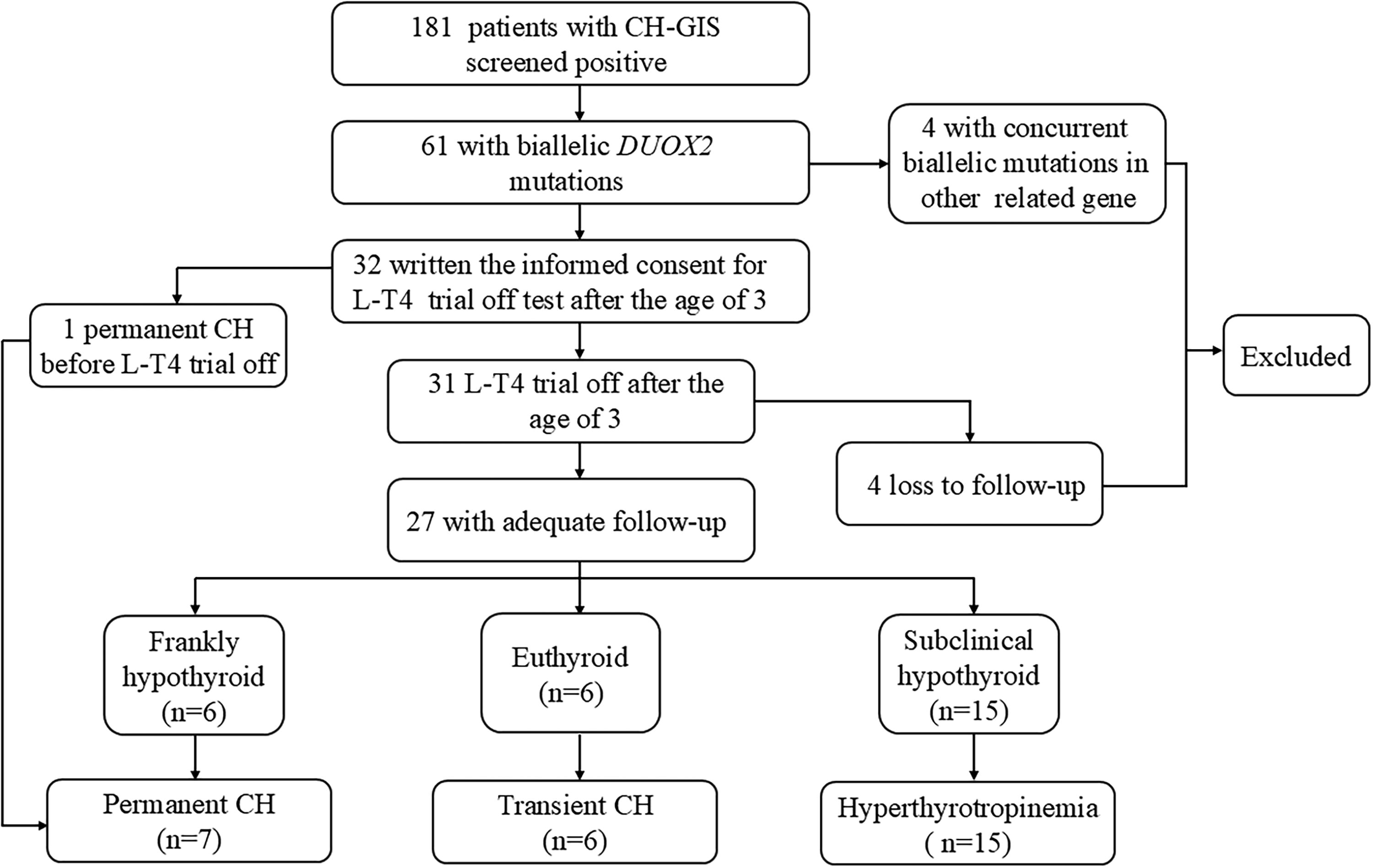
The flowchart of patients included in the study and classification of different forms of CH. CH, congenital hypothyroidism; GIS, gland in situ; LT4, levothyroxine.
Clinical reevaluation and long-term follow-up
After 3 years of age, the parents of 32 patients have written the informed consent about LT4 trial off test before clinical reevaluation. Out of these 32 patients, one patient was diagnosed with PCH because the TSH level was >10 µIU/mL in one test during the first year of life during LT4 treatment. This CH patient was characterized by severe growth and mental retardation; the patient’s height was three standard deviations less than the mean, and intelligence was lower than that of 80.9% of children within the reference age. Hence, the LT4 withdrawal trial was not performed for this patient. After 3 years of age, LT4 therapy was suspended in 31 patients. Of which, 4 with an insufficient time of follow-up after suspension of LT4 therapy (i.e., <6 months) were excluded. The remaining 27 patients were with adequate follow-up after suspension of LT4 therapy (at least 6 months). These patients were reevaluated at 1, 3, and 6 months after LT4 withdrawal (Fig. 1). PCH was defined as a TSH level >10 µIU/mL in at least one test. TCH was defined when thyroid function tests were normal at 1, 3, and 6 months after LT4 withdrawal. Hyperthyrotropinemia was defined as elevated TSH levels but <10 µIU/mL in two or more consecutive tests with normal fT4 levels at 1, 3, and 6 months after LT4 withdrawal.
For patients diagnosed with PCH after LT4 withdrawal, LT4 treatment was restarted immediately. For patients with TCH and hyperthyrotropinemia, LT4 treatment was suspended during the follow-up period. Age, body height, body weight, thyroid function, thyroglobulin (Tg) levels, LT4 dosage (for PCH), and thyroid ultrasound were regularly recorded every 3–6 months. The need for resumption of LT4 treatment was evaluated based on their clinical manifestations individually.
Medical records
During LT4 treatment, periodic follow-up and drug dose adjustments were performed for all patients according to the current guidelines. 3,5 Perinatal data (sex, gestational age, birth weight, and TSH level during newborn screening) and family history of CH, genetic information, or thyroid disease were obtained by thoroughly reviewing the medical records. Age, height, weight, thyroid function tests, and LT4 dose during the treatment period and LT4 withdrawal trial were recorded (Supplementary Table S1 and Supplementary Table S2). Thyroid ultrasonography or technetium-99 (Tc-99m) thyroid scan was performed at a local center during diagnosis or follow-up. Thyroid lobe volume was calculated using the following formula: 0.526 × length (cm) × width (cm) × thickness (cm). The total thyroid volume was the sum of the volumes of the two lobes. Goiter was defined as the thyroid size exceeding the upper limit of normal (ULN) reported in the literature. 14 Thyroid ultrasonography data and changes in thyroid size over time have been provided in the Supplementary Table S3.
Statistical analysis
Statistical analyses were performed using IBM Statistical Product and Service Solutions, version 23.0 (IBM Co., Armonk, NY, USA). p-Values <0.05 were considered statistically significant. Categorical variables are presented as frequencies (n) and percentages (%), and numeric variables are presented as medians (minimum to maximum ranges) if numeric variables showed a non-normal distribution or as means ± standard deviations if they showed a normal distribution. The Kolmogorov–Smirnov test was used to evaluate normality. The chi-squared test or Fisher’s exact test was used to compare the categorical variables. One-way analysis of variance was used to compare continuous variables among TCH, PCH, and hyperthyrotropinemia groups.
Results
Clinical outcomes of patients with CH and DUOX2 biallelic mutations
One patient was diagnosed with PCH before LT4 withdrawal trial as described above. Of 27 patients with adequate follow-up after suspension of LT4 therapy (the median duration of follow-up was 17.5 months [interquartile range: 8.5, 29.25]), 6 patients presented serum TSH concentration >10 µIU/mL at least once within 6 months of withdrawal, and they were classified as PCH. Six patients were euthyroid within 6 months of withdrawal and were classified as TCH. However, 15 showed serum TSH concentrations above the upper limit of the reference interval (ULN) but <10 µIU/mL at least once within 6 months after LT4 withdrawal, and they were classified as hyperthyrotropinemia. Finally, a total of 28 patients, including 7 PCH (7/28, 25%), 6 TCH (6/28, 21.4%), and 15 hyperthyrotropinemia (15/28, 53.6%), were enrolled in the subsequent analysis (Fig. 1).
All the patients underwent routine thyroid ultrasound examinations to evaluate the position and size of the thyroid gland at reevaluation. Of which, 12 patients also performed Tc-99m thyroid scan before LT4 withdrawal. The results showed that all patients were of in situ thyroid with normal size before LT4 withdrawal. Tc-99m pertechnetate thyroid uptake rate in 10 patients was normal; 2 were slightly declined and returned to normal after LT4 withdrawal.
Molecular analysis of patients with CH and DUOX2 biallelic mutations
In total, 34 DUOX2 variants were determined in these 28 patients. Based on The American College of Medical Genetics and Genomics (ACMC) guidelines, 15,16 12 were pathogenic variants, 15 were likely pathogenic variants, and 7 variants were uncertain significance (Supplementary Table S2). Out of the 28 patients with CH who carried DUOX2 biallelic mutations, 11 patients also harbored other variants, spanning 11 CH-related genes: DUOX1, DUOXA1, DUOXA2, NKX2-1, PAX8, TG, THRA, FOXE1, DIO1, TRHR, and GNAS. Expect DUOX2 variants; four patients carried one variant in other genes related to CH (2 in TCH, 1 in PCH, and 1 in hyperthyrotropinemia), and seven patients carried more than two variants in other genes related to CH (1 in TCH, 2 in PCH, and 4 in hyperthyrotropinemia) (Supplementary Table S1).
Clinical parameters in patients with CH and DUOX2 biallelic mutations before reevaluation
Comparison of clinical data among the PCH, TCH, and hyperthyrotropinemia groups revealed no significant differences in birth weight (p = 0.279), gestational age (p = 0.359), and fT4 (p = 0.340) levels at diagnosis. Heel blood TSH (NBS) and TSH at diagnosis trended higher in PCH/hyperthyrotropinemia versus TCH, but differences were not significant (p = 0.499; p = 0.877) (Table 1). No relationship was found between DUOX2 residual enzymatic activity and clinical outcomes. Unexpectedly, DUOX2 enzymatic activity was significantly higher in hyperthyrotropinemia versus TCH (33.43 ± 9.91 vs. 21.60 ± 5.59, p = 0.027) (Table 1).
Clinical and Biochemical Features at Diagnosis
Categorical variables are presented as frequencies (n) and percentages (%); numeric variables are presented as median (range) if numeric variables were abnormal distribution or mean ± SD (standard deviation) if numeric variables were normal distribution. p-Value was calculated among three groups by one-way analysis of variance (ANOVA).
Significant difference for DUOX2 enzymatic activity between patients with transient CH and hyperthyrotropinemia (p = 0.028).
CH, congenital hypothyroidism; F, female; fT4, free T4; M, male; TSH, thyrotropin.
During the treatment period, initial LT4 dose and average dose at 1 year did not differ significantly between groups (p = 0.629; p = 0.273); however, the average doses of LT4 at 2 and 3 years of age were significantly higher in PCH versus TCH (2.74 ± 0.53 vs. 1.55 ± 0.41, p = 0.027; 2.84 ± 0.90 vs. 1.11 ± 0.46, p = 0.003) (Table 2). LT4 dose decreased with age in TCH/hyperthyrotropinemia but remained stable in PCH. Furthermore, the average LT4 dose at 3 years of age was significantly higher in PCH versus hyperthyrotropinemia (1.85 ± 0.72 vs. 1.11 ± 0.46, p = 0.027) (Table 2).
Clinical and Biochemical Features of patients with CH During Treatment with LT4 Before Reevaluation
Data are presented as mean ± SD (standard deviation). p-Value was calculated among three groups by one-way analysis of variance (ANOVA).
Significant difference between patients with PCH and hyperthyrotropinemia (p < 0.05).
Significant difference between patients with PCH and TCH (p < 0.05).
LT4, levothyroxine; PCH, permanent congenital hypothyroidism; TCH, transient congenital hypothyroidism.
Patients after 3 years of age were reevaluated following the withdrawal of LT4 replacement therapy. The average ages of LT4 withdrawal in the PCH, hyperthyrotropinemia, and TCH groups were 72, 64, and 49 months, respectively (Table 2). TSH and fT4 pre-withdrawal did not differ significantly between groups (p = 0.204; p = 0.575) (Table 2). However, the LT4 dose pre-withdrawal was significantly higher in PCH versus TCH (2.48 ± 1.14 vs. 1.18 ± 0.62, p = 0.025) and showed a declining trend among three groups (PCH > hyperthyrotropinemia > TCH) (Table 2).
Long-term follow-up for TCH and hyperthyrotropinemia in patients with CH and DUOX2 biallelic mutations
Long-term follow-up (more than 12 months) of patients with CH and biallelic DUOX2 mutations showed no significant differences in weight/height gain rates between the PCH, hyperthyrotropinemia, and TCH groups (p = 0.503; p = 0.757) (Table 3). Median follow-up times after LT4 withdrawal were 24, 12, and 42 months, respectively (Table 3). The mean TSH level within the first six months post-withdrawal and after one year of withdrawal were both significantly higher in the hyperthyrotropinemia group than in the TCH group (5.98 ± 1.42 vs. 2.66 ± 0.86, p < 0.001; 4.86 ± 1.39 vs. 2.38 ± 0.80, p = 0.005) (Table 3). Mean TSH levels after 1 year declined compared with the first 6 months in both the hyperthyrotropinemia (5.98 ± 1.42 vs. 4.86 ± 1.39 µIU/mL; p = 0.06) and TCH groups (2.66 ± 0.86 vs. 2.39 ± 0.81 µIU/mL; p = 0.60). (Table 3). All patients with TCH maintained normal thyroid function during follow-up. Of five patients followed for more than 1 year, two had persistently elevated Tg (Fig. 2). Of nine patients with hyperthyrotropinemia followed for more than 1 year, four had persistently elevated Tg and TSH (>reference interval but <10 µIU/mL), four normalized both, and one had persistently elevated TSH (Tg not tested) (Fig. 2) (Supplementary Table S3). This finding indicated that half of patients with hyperthyrotropinemia may normalize thyroid function over time by thyroid compensatory hyperplasia.
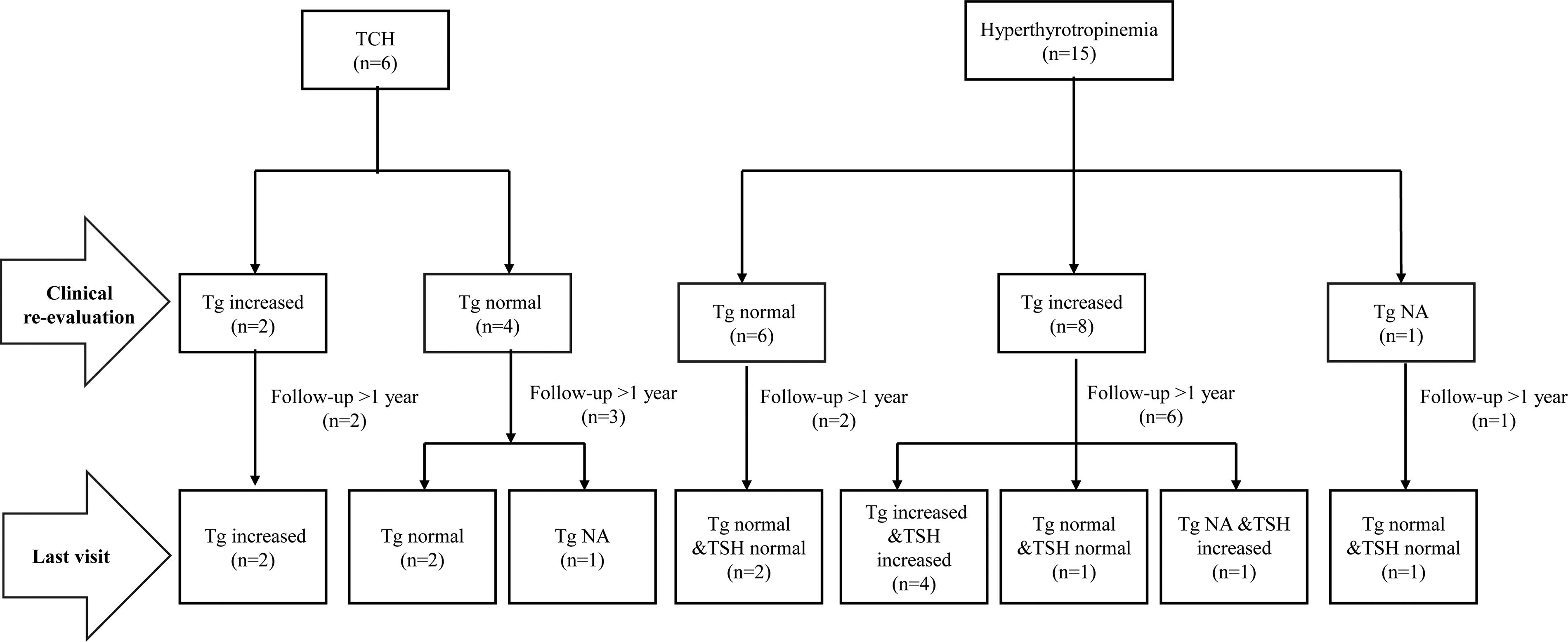
Diagram of the clinical feature of TCH (on the left side) and hyperthyrotropinemia (on the right side). Tg, thyroglobulin.
Clinical and Biochemical Features During Long-Term Follow-up after LT4 Withdrawal
Date was presented as median (minimum to maximum range) if numeric variables were abnormal distribution or mean ± SD (standard deviation) if numeric variables were normal distribution. p-Value was calculated among three groups by one-way analysis of variance (ANOVA).
Significant difference between patients with hyperthyrotropinemia and transient CH (p < 0.05).
Significant difference between patients with permanent CH and transient CH (p < 0.05).
Patient 10, a male patient with CH and DUOX2 biallelic mutations, had discontinued LT4 therapy at 3.5 years of age. This patient had mildly elevated TSH (<10 µIU/mL) and rising Tg until 33 months post-withdrawal, when TSH reached 10.68 µIU/mL and Tg 91.38 ng/mL. Thyroid volume was at the 84th percentile. Without restarting LT4, TSH decreased to 5.55 µIU/mL within one month, coinciding with significant thyroid volume increase to 8.21 mL (>24% above ULN). Given normal growth (40 kg, 140 cm at age 8), LT4 was not restarted, but close monitoring continues (Supplementary Fig. S1).
Discussion
In this prospective study, we reevaluated the clinical outcomes of a cohort of 28 patients with CH and DUOX2 biallelic mutations at 3 years of age and established a long-term follow-up to track the clinical course of these patients. After clinical reassessment, 25% (7/28) of the patients were confirmed to have PCH, 21.4% (6/28) had TCH, and 53.6% (15/28) presented with hyperthyrotropinemia. This finding has an important prognostic value because it confirms that at least some cases of CH caused by DUOX2 biallelic mutations could discontinue LT4 replacement therapy after the age of three, which was different from the previous view that CH caused by dyshormonogenesis required lifelong treatment with LT4. 1
Several studies have investigated the predictors of TCH, such as low birth weight, premature birth, iodine insufficiency, and iodine exposure during pregnancy. 17 –19 Delayed TSH elevation is common in preterm infants, and these patients generally have TCH. 20 Out of 28 patients with CH and DUOX2 biallelic mutations in our study, only one was a preterm baby; however, the TSH level in that patient was elevated during newborn screening, and they were diagnosed with PCH because of the severe growth and mental retardation observed at 3.5 years of age. In addition, the LT4 dose in patients with CH during replacement therapy is the best predictor of PCH and TCH. 21 –24 Although the required dose decreases gradually with age in children with TCH, it increases gradually in those with PCH. 25 –28 The similar results were found in our study, which showed significantly lower LT4 dose in the TCH group than in the PCH group at two and three years of age. Whether the laboratory findings can predict TCH remains controversial. Previous studies have suggested that children with TCH have significantly lower TSH levels at onset than those with PCH. 17 However, other studies have reported that the fT4 and TSH levels during onset did not differ between patients with TCH and PCH. 7,29 In our study, the serum TSH levels during newborn screening and diagnosis showed an incremental trend in the PCH group compared with the TCH group, without a significant difference. These findings suggest that the serum TSH level at the onset is not superior to the LT4 dose in predicting PCH and TCH.
Based on previous guidelines, classifying a form of CH as permanent or transient is through reevaluation of the thyroid axis, which should normally take place after the age of three years. 3 While recent studies have recommended earlier reevaluation by two years and even earlier if infants with CH requiring lower LT4 doses or in most very low-birth-weight infants who were diagnosed as mild form of CH. 30,31 In the recent European Society for Pediatric Endocrinology and the European Society for Endocrinology guidelines, the reevaluation for CH is suggested at six months in patients with no PCH diagnosis and an in situ gland who needs LT4 dose of <3 µg/kg/day. 5 In our study, the pathogenesis of CH was definite in these enrolled patients who carried DUOX2 biallelic mutations. Although the maintained LT4 dose was less than 3 µg/kg/day in most patients, it may not be sufficient to predict their clinical outcome because their clinical phenotype showed a great heterogeneity. It is well known that the most thyroxine-dependent brain maturation has occurred during the first three years of life. 32 Therefore, out of caution, we performed reevaluation after three years of age for these patients with CH, just as the latest American Pediatric Academy guidelines suggested. 4
It has been reported that the clinical phenotypes of patients with DUOX2 biallelic mutations show great variability. Our previous study had indicated that the DUOX2 enzymatic activity was closely related with clinical phenotypes at diagnose. 33 Therefore, we assumed that there may be certain correlation between clinical outcome and DUOX2 enzymatic activity in patients with CH with DUOX2 biallelic mutations. Theoretically, the lower the DUOX2 enzymatic activity, the more severe the phenotype. The DUOX2 enzymatic activities in PCH should be lower than those of hyperthyrotropinemia group, the latter also lower than those of TCH group, as speculated. However, in our study, the highest DUOX2 enzymatic activity in these three groups was hyperthyrotropinemia, followed by PCH and TCH. No relationship was observed between the DUOX2 residual enzymatic activity and the clinical outcomes in patients with DUOX2 biallelic mutations after withdrawal of LT4. While most patients who discontinued LT4 therapy had residual DUOX2 activity ≥22% (TCH: 66.7% [4/6]; hyperthyrotropinemia: 93.3% [14/15]). Therefore, we hypothesized that only when the residual enzyme activity exceeds a certain level in patients with DUOX2 biallelic mutations, it might manifest as TCH or hyperthyrotropinemia by compensating for the deficiency of thyroid hormone synthesis in single thyrocytes through goiter after LT4 withdrawal. Thus, the occurrence of hyperthyrotropinemia and TCH in patients with DUOX2 biallelic mutations may partially be explained by DUOX2 enzymatic activity, but it could not account for the clinical outcomes of PCH. However, compensatory thyroid hyperplasia may be affected by several factors, such as TSH concentration, iodine intake, and duration of drug withdrawal. In patients with PCH who retain >22% residual DUOX2 enzyme activity, a short period of LT4 withdrawal overwhelms the compensatory capacity of thyroid hyperplasia, resulting in inadequate thyroid function. This aligns with our finding that most patients with PCH discontinued therapy for less than three months. Other determinants or modifiers beyond DUOX2 enzymatic activity may also have influenced the variable phenotypes, such as other associated gene variants and iodine intake.
Kara et al. have revealed the importance of genetics in the management of patients with CH, which can change diagnosis and treatment decisions in a small proportion of children with CH. 9 It is well known that DUOX1 activity can compensate for the DUOX2 defect. 34 In our study, seven patients carried DUOX1 or DUOXA1 mutation. Of which, only one patient (1/6, 16.7%) carried DUOXA1 variant in TCH group, while four patients (4/15, 27%) in hyperthyrotropinemia carried DUOX1 or DUOXA1 variants. Moreover, only one patient (1/6, 16.7%) carried variant in other genes related to thyroid dyshormonogenesis in TCH group, while in PCH and hyperthyrotropinemia group, the proportion was 28.6% patients (2/7) and 33.3% (5/15), respectively. In addition, other variants in the genes encoding transcription factors (FOXE1, PAX8, NKX2-1) and TSH signaling molecular (GNAS) or thyroid hormone receptor (THRA) were also observed in the patients with CH with DUOX2 biallelic mutations. Although variants in other thyroid genes showed similar distribution across groups, their potential effect on disease severity in patients with CH with biallelic DUOX2 mutations should be considered.
Environmental factor, especially iodine nutritional status, acts as a disease modifier of genetic CH. 19,35 There are reports supporting that individuals with the same DUOX2 variants may show different clinical presentations depending on iodine exposure. 36 Abnormal urinary iodine levels with high Tg concentrations were associated with lower LT4 dose requirements. 37 In our study, urinary iodine was not available; whether there was a concomitant iodine deficiency in patients with elevated TSH and Tg should be further investigated.
The management of isolated TSH elevation (also known as subclinical hypothyroidism [SCH]) was controversy, but today there are generally accepted recommendations and consensus points. 38,39 Accordingly, treatment with LT4 should be routinely applied in patients with severe SCH (TSH >10 µIU/mL). In patients with mild SCH (TSH: 4.5–10 µIU/mL), the management varies depending on condition: if the patient has goiter or symptoms of hypothyroidism, treatment is recommended; otherwise, monitoring without treatment is preferred. 39 While standard subclinical hypothyroidism (SCH) management is established, mild hyperthyrotropinemia (TSH 4.5–10 µIU/mL) in patients with DUOX2 biallelic mutation post-LT4 withdrawal requires specialized consideration. Due to the insufficiency of DUOX2 activity in individual thyrocytes, thyroid hyperplasia (increased cell number) is necessary to maintain euthyroidism post-LT4 withdrawal. Therefore, for patients with DUOX2 biallelic mutations and mild hyperthyrotropinemia, LT4 therapy was not restarted despite mild thyroid hyperplasia or rising Tg levels, as in this study. Instead, long-term monitoring was implemented to prevent excessive thyroid enlargement. If thyroid sizes were increased continuously and obviously above the ULN, LT4 retreatment should be restarted.
Even for patients with TCH, although the majority of patients with TCH sustained euthyroid after LT4 withdrawal, the thyroid function in a minority of patients may get worse over a long-term follow-up despite an initial normalization within the earlier years after LT4 withdrawal. 6 However, to date, no recommendations are available about the specific follow-up time based on the guidelines. Considering the required thyroid hormone is increased at particular periods such as puberty and pregnancy, it is very reasonable to periodically evaluate the thyroid function of patients with both hyperthyrotropinemia and TCH after LT4 withdrawal until the end of linear growth or during pregnancy to avoid the adverse effect of thyroid hormone insufficiency on these patients.
This study has limitations due to its small sample size; the clinical outcome (proportions of PCH, TCH, and hyperthyrotropinemia) for patients with CH and DUOX2 biallelic mutations after LT4 withdrawal needs to be verified in a larger cohort in the future. In addition, although DUOX2 is an important gene related to thyroid hormone synthesis, we did not perform a perchlorate discharge test during reevaluation because the medicinal use of this compound was not available in China. Research with a larger sample size is required to further elucidate PCH and TCH in patients with DUOX2 biallelic mutations.
Conclusions
The results of this study highlight the clinical evolution of patients with CH and DUOX2 biallelic mutations, which showed only 25% patients need lifelong therapy, while 75% patients can stop levothyroxine therapy after three years of age. However, after levothyroxine withdrawal, serum thyroglobulin levels and thyroid size increased in most patients, especially in the hyperthyrotropinemia group. Therefore, long-term follow-up are required to determine the outcome of patients with CH, especially for patients with hyperthyrotropinemia, to avoid overtreatment or negative consequences for development due to inadequate treatment.
Footnotes
Acknowledgment
The authors are grateful to all patients, clinicians, and hospital staff participating in the study.
Authors’ Contributions
H.-D.S. conceived and designed the project. H.-D.S., S.-X.Z., and F.S. contribute to the project management. F.S., J.-P.W., and W.-H.D. contributed to methodology, analysis, investigation, and data curation. J.N., H.-Y.Z., C.-Y.W., Q.-Y.Z., R.-M.Y., Y.F., X.-S.L., and F.-Y.W. took part in the methodology and investigation data curation. H.-D.S., S.-X.Z., F.S., B.H., X.-S.L., F.C., and L.L. contributed to funding acquisition. F.S. and H.-D.S. wrote the article. The article was read by all authors and approved for submission.
Author Disclosure Statement
The authors have nothing to declare.
Funding Information
This work was supported by National Natural Science Foundation of China (82200874, 82070816, 82070889, 82300879, 82270826, 82200873, 82103788, and 82170802), Innovative Research Team of High-Level Local Universities in Shanghai (SHSMU-ZDCX20210901 and SHSMU-ZDCX20212501), Natural Science Foundation of Shanghai (22ZR1436600 and 24ZR1443100), Top Talent Support Program of Minhang Hospital, Fudan University (MZBJ06), and Natural Science Foundation of Fujian Province (2022J01438).
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.