
Abstract
Background:
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disorder characterized by progressive cyst development and renal dysfunction. While thyroid hormones (THs) are known to regulate key pathways in various kidney diseases, their role in ADPKD pathobiology and therapeutic potential remains unexplored. Here, we aimed to elucidate the role of THs in ADPKD and evaluate whether their pharmacological modulation could serve as a therapeutic strategy.
Methods:
Patient-derived renal epithelial cells were used to engineer 3D polycystic tubules and to test the anti-cystogenic effects of THs and their analogs. The therapeutic efficacy of thyroxine (T4) in reducing cyst formation and delaying disease progression was assessed in vivo using PCK rats, an animal model of ADPKD. Lastly, serum THs levels were measured in 90 ADPKD patients enrolled in the REORIENTED clinical study and correlated with estimated glomerular filtration rate to explore their clinical relevance (Clinical Trial Gov NCT 05646420).
Results:
Mechanistically, thyroxine inhibits cyst growth by modulating proliferative, metabolic and ferroptotic pathways through αvβ3 integrin binding. In PCK rats, an animal model of ADPKD, T4 administration decreased kidney weight and significantly reduced macrocystic area (%, Vehicle 9.255 ± 2.654 vs. T4 1.945 ± 0.850, p < 0.05). Clinical data from ADPKD patients showed that altered TH serum levels correlate with disease severity: in the overall population of the study reverse triiodothyronine (rT3, a T3′s metabolite) levels inversely correlate with renal function (R2 = 0.159, r = −0.397, p < 0.001), while free triiodothyronine (fT3) levels show a positive correlation (R2 = 0.110, r = 0.332, p < 0.01).
Conclusions:
This study reveals that THs contribute to ADPKD progression and identifies them as potential prognostic and therapeutic agents. By modulating multiple pathogenic pathways, THs may offer a novel, multi-targeted approach to reduce cyst growth and preserve renal function. These findings further support the development of personalized, hormone-based treatments and more refined stratification in the clinical management of ADPKD.
Keywords
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common rare monogenic disorder affecting humans, with a prevalence of 1/1000 individuals. 1 It is characterized by the development of multiple abnormal fluid-filled cysts originating from renal tubules, which disrupt kidney architecture and function. 2
Current therapeutic options are limited to anti-hypertensive therapies 3 and drugs that slow cyst growth, such as Tolvaptan and Octreotide-LAR. 4 While these treatments can slow disease progression, they present side effects5–7 and a significant number of ADPKD patients still progress to end-stage kidney disease. There is, therefore, an urgent need to identify novel mechanisms driving disease pathogenesis and to develop more effective therapeutic strategies.
ADPKD is primarily caused by loss-of-function mutations in genes encoding for polycystins PC1 and PC2, leading to epithelial cell dedifferentiation, uncontrolled proliferation, abnormal fluid secretion, and metabolic remodeling.8,9 The complexity and interdependence of these processes suggest that pleiotropic regulators may play a central role in disease progression.
Thyroid hormones (THs) are key regulators of cellular metabolism, growth, differentiation, and survival, and therefore represent promising candidates.10–13 TH signaling operates through genomic pathways mediated by nuclear receptors and non-genomic pathways involving αvβ3 integrin,14,15 which controls key proliferative and survival pathways. Importantly, TH derivatives such as triiodothyroacetic acid (TRIAC) and tetraiodothyroacetic acid (TETRAC) can differentially modulate αvβ3 integrin signaling, exerting actions that diverge from or oppose those of triiodothyronine (T3) and thyroxine (T4) 16 , the two major active forms of THs.
Consistent with their role in cellular homeostasis, THs regulate cellular energetics, a central hallmark of ADPKD pathobiology. Cystic cells display enhanced glycolysis and glutamine metabolism to sustain proliferation and survival.17,18 These metabolic alterations directly impact cellular redox balance and glutathione metabolism, key determinants of ferroptosis susceptibility.19,20 Ferroptosis—an iron-dependent form of regulated cell death driven by lipid peroxidation—has recently been implicated in disease progression. 21 Given the central role of THs in metabolic and redox regulation, they may critically modulate these pathogenic pathways.
Consistently, reduced TH levels have been associated with cellular dedifferentiation and reactivation of cell cycle in the heart and kidney,10,12,13 as well as cyst formation in ovaries, 22 while TH supplementation has been shown to reverse these alterations.10,13,22,23 Remarkably, approximately 80% of chronic kidney disease (CKD) patients display altered THs profiles, 24 including those with ADPKD; 25 however, the role of TH signaling in the pathobiology of ADPKD remains largely unexplored.
Based on this rationale, we aimed to elucidate the role of TH signaling in ADPKD pathobiology and to assess its potential as a therapeutic strategy to reduce cyst burden and slow disease progression.
Materials and Methods
Cystogenesis assay in ADPKD patient-derived polycystic tubules
Primary single cyst-derived huADPKD cells were cultured to obtain tubules as previously described.26,27 Triiodothyronine and thyroxine concentrations were selected based on previous studies demonstrating effective and non-toxic activation of TH signaling in vitro.13,23,28,29 Cells and/or tubules were treated as follows: T3 (0.5 µM), T4 (0.1 µM), TRIAC (25 µM), TETRAC (25 µM), DBD (0.5 µM), LM609 (2.5 µg/ml), UO126 (0.1 µM), and CB839 (1 µM). Immunofluorescence and western blot were performed as previously described.13,27
Seahorse XF real-time ATP rate assay
To measure glycolysis and mitochondrial rate, cells were plated in a Seahorse 24-well plate and incubated in standard condition (5% CO2 at 37°C) overnight. The following day, cells were treated with T4 alone or in combination with LM609 or CB839 for 24 hours (Supplementary Table S1). At the end of the experiment, the Agilent Seahorse XF real-time ATP rate Assay was performed according to the manufacturer’s instructions. Reads were normalized on cell lysates.
Targeted metabolomics
Frozen samples were homogenized with 10 µL/mg of extraction solvent (85:15 MeOH/H2O), incubated at −80°C for 20 minutes and subsequently centrifuged for 15 min at 13,000 g. Supernatants were collected and 20 µL of each extract was used for targeted metabolomic analysis. A targeted quantitative AbsoluteIDQ® MxP500 Quant kit (Biocrates) was performed following the manufacturer’s instructions. Metabolomics workflow management and data processing were carried out using the WebIDQ software (Biocrates).
Real-time PCR
Total RNA was extracted from huADPKD cells with Trizol reagent following the manufacturer’s instructions. Real-time PCR was performed as previously described. 30
Fe2+ and lipid peroxidation measurements
To measure intracellular Fe2+ and lipid peroxidation, huADPKD cells were seeded and treated for 40 hours with T4. At the end of the experiment, Ferrous Iron kit (E-BC-K881-M) and Bodipy 581/591 C11 (HY-D1301) assay were performed following manufacturers’ instructions.
Animals
Female PCK rats (n = 7/group) were randomly divided into two groups and treated daily by gavage from 4 to 11 weeks of age with vehicle (water), or L-thyroxine (T4, 10 µg/kg). Sample size and T4 dose were selected based on previous studies, showing TH beneficial effect without major side effects.23,31 Age- and sex-matched Sprague-Dawley (SD) rats (n = 5) were used as control.
Clinical study
REORIENTED was an exploratory, biochemical study (Clinical Trial Gov NCT05646420) with a prospective design that included patients with ADPKD at different CKD stages, ranging from normal kidney function to stage IV CKD.
Statistical analysis
Statistical analyses were performed using the GraphPad Prism software. The two-tailed Student’s t-test or one-way ANOVA with Tukey’s or Bonferroni multiple comparisons test were used as appropriate. The correlation between serum T4 levels and macrocystic area in PCK rats were determined by linear regression while the correlation between serum THs levels and eGFR in ADPKD patients were analyzed by both linear regression and Pearson correlation. Significant differences were considered when p < 0.05.
Detailed methods of all sections are reported in supplemental information
Institutional review board approvals
All procedures involving animals were performed in accordance with institutional guidelines in compliance with national (D.L.n.26, March 4, 2014) and international laws and policies (Directive 2010/63/EU) and were approved by the Institutional Animal Care and Use Committees of the Istituto di Ricerche Farmacologiche Mario Negri IRCCS. The REORIENTED clinical study was registered at Clinical Trial Gov NCT05646420. Institutional review board approval was obtained by the Ethical Committee of the ASST Papa Giovanni XXIII (Reg. 230/22) and written informed consent was obtained from each patient included in the study. The study was completed in accordance with the Declaration of Helsinki as revised in 2013.
Results
T4 inhibits cyst growth in vitro
To test the potential anti-cystogenic effects of THs and their derivatives15,16,22—TRIAC (a T3 metabolite) and TETRAC (a deaminated analogue of T4)—in vitro, we treated engineered ADPKD patient-derived polycystic tubules.26,27 Notably, T3 and T4 demonstrated marked anti-cystogenic properties (CTRL 81.69 ± 3.32 vs. T3 48.40 ± 3.44 cysts/mm2, p < 0.0001; CTRL 81.69 ± 3.32 vs T4 45.52 ± 2.97 cysts/mm2 p < 0.0001; Fig. 1A,B), significantly reducing cyst number and size, whereas TRIAC and TETRAC exhibited a pro-cystogenic effect (CTRL 81.69 ± 3.32 vs TRIAC 101.1 ± 3.90 cysts/mm2 p < 0.01; CTRL 81.69 ± 3.32 vs. TETRAC, 113.2 ± 8.26 cysts/mm2, p < 0.0001; Fig. 1A,B).
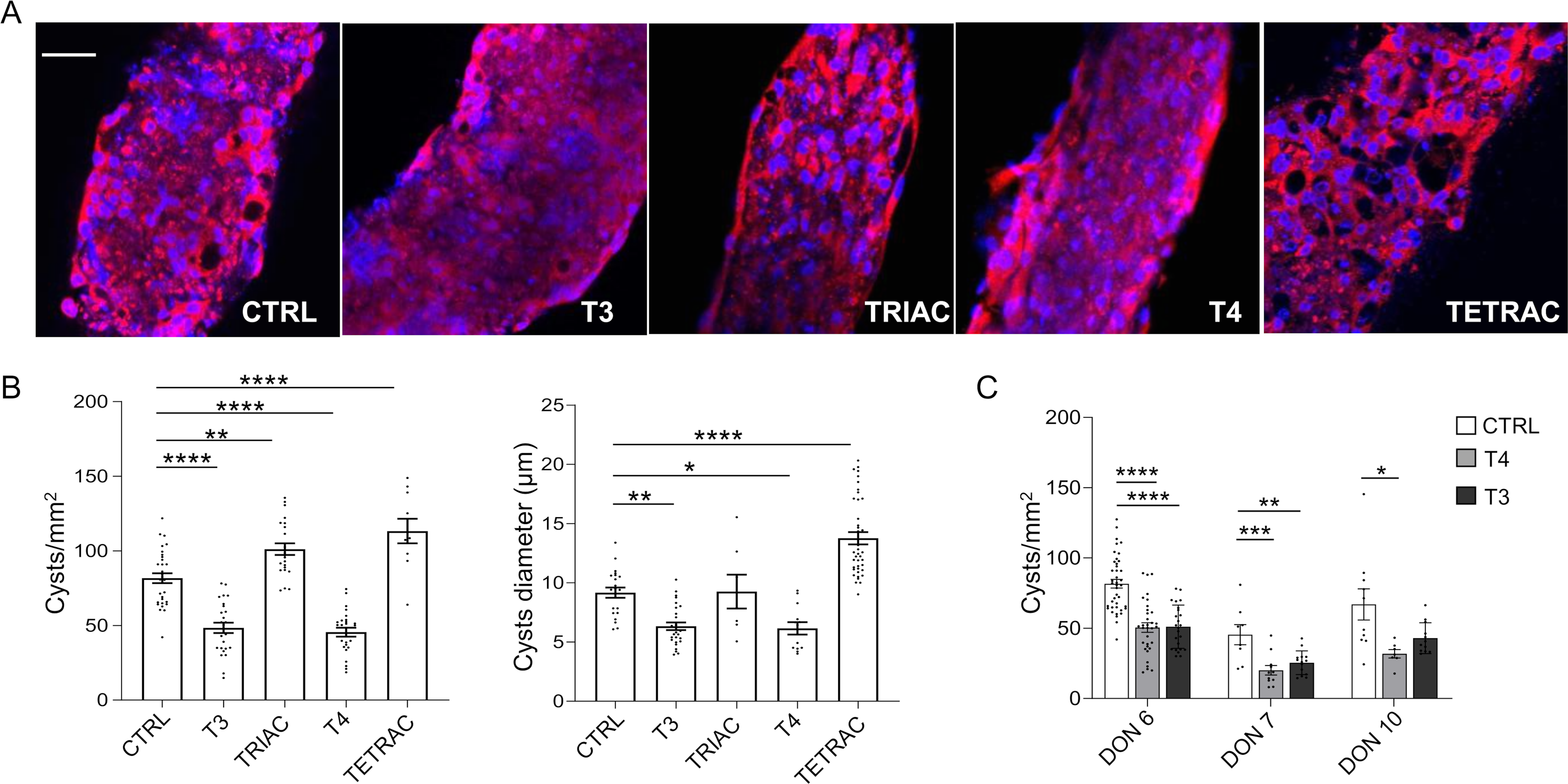
Effect of THs treatment on cyst number and size in patient-derived polycystic tubules.
Consistency and reproducibility of THs anti-cystogenic effect were evaluated on polycystic tubules from three different ADPKD patients. Both T3 and T4 demonstrated anti-cystogenic properties, with T4 showing more consistent effects across patient-derived samples (Fig. 1C).
The in vitro anti-cystogenic effect of T4 is mediated by integrin αvβ3 binding and MAPK cascade inhibition
To assess whether the anti-cystogenic effects of THs were mediated by genomic or non-genomic pathway,14–16 we treated patient-derived polycystic tubules with debutyl-dronedarone (DBD), a competitive inhibitor of T3 binding to TRα, 32 the nuclear TH receptor that controls proliferation in various tissues,33–35 or with the monoclonal antibody LM609, which sterically hinders the T4 binding site on αvβ3 integrin. 36 Our findings revealed that DBD treatment did not affect T4 efficacy (CTRL 79.37 ± 3.166 vs. T4 50.73 ± 3.075 cysts/mm2 p < 0.0001; CTRL 79.37 ± 3.166 vs. DBD + T4 51.33 ± 5.472 cysts/mm2, p < 0.001; T4 50.73 ± 3.075 vs. DBD + T4 51.33 ± 5.472 cysts/mm2, ns, Fig. 2A), whereas LM609 treatment abrogated it (CTRL 80.31 ± 3.317 vs. T4, 51.03 ± 3.315 cysts/mm2 p < 0.0001; CTRL 80.31 ± 3.317 vs. LM609 + T4 71.83 ± 9.400 cysts/mm2, ns; T4 51.03 ± 3.315 vs. LM609 + T4 71.83 ± 9.400 cysts/mm2, p < 0.05, Fig. 2B), confirming that T4’s anti-cystogenic effect is, at least partially, mediated through its binding to αvβ3 integrin.

T4 treatment exhibits a strong anti-cystogenic effect in patient-derived polycystic tubules through the non-genomic pathway. Immunofluorescence for E-cadherin (red) on patient-derived polycystic tubules treated with T4 and DBD
Activation of integrins is known to influence key signaling pathways involved in cell proliferation, survival, and differentiation. 37 To explore whether T4’s anti-cystogenic effect occurs via suppression of proliferative signaling, we analyzed levels of total and phosphorylated extracellular signal-regulated kinase (ERK and p-ERK, respectively), a downstream effector of the MAPK cascade. Interestingly, T4 treatment significantly reduced the p-ERK/ERK ratio compared with controls (CTRL 2.054 ± 0.334 vs. T4 0.314 ± 0.034 p < 0.01, Fig. 2C), an effect further corroborated by p-ERK staining in cells from patients’ cysts (Fig. 2D). Notably, pretreatment with LM609 attenuated this reduction (CTRL 58.65 ± 2.824 vs. T4 29.51 ± 3.112 p-ERK+-cells, p < 0.0001; CTRL 58.65 ± 2.824 vs. LM609 + T4 43.25 ± 2.722 p-ERK+-cells, p < 0.001; T4 29.51 ± 3.112 vs. LM609 + T4 43.25 ± 2.722 p-ERK+-cells, p < 0.01, Fig. 2D), further supporting the role of αvβ3-mediated signaling in T4’s mechanism of action. Finally, to better elucidate the role of ERK in cyst induction and progression, we treated patient-derived tubules with UO126, a specific ERK inhibitor. This treatment significantly reduced cyst formation and growth, supporting the notion that T4’s effect may be mediated through ERK downregulation (CTRL 81.69 ± 3.094 vs. T4, 51.03 ± 3.315 cysts/mm2 p < 0.0001; CTRL 80.31 ± 3.317 vs. UO126 46.98 ± 3.785 cysts/mm2, ns; T4 51.03 ± 3.315 vs. UO126 46.98 ± 3.785 cysts/mm2, ns, Fig. 2E).
T4 and cellular energetics
To study the effect of T4 on human cyst cell energetics, 17 we analyzed both the glycolytic and OXPHOS pathways and found that T4 treatment increases total energy production compared with controls (maximal glycolytic rate of CTRL 1.089 ± 0.0819 vs. T4 2.059 ± 0.092 pmolH+/min, p < 0.0001; basal OXPHOS of CTRL 5.277 ± 0.402 vs. T4 8.814 ± 0.646 pmolO2/min, p < 0.0001; Fig. 3A,B) and that this effect is independent of integrin binding (Supplementary Fig. S1). To further dissect the molecular mechanisms driving this metabolic stimulation, we blocked glutamine usage, which plays a crucial role in ADPKD metabolism 17 and is controlled by TH signaling, 38 with the glutaminase inhibitor CB839. The results showed that CB839 treatment completely inhibits the effect of T4 on OXPHOS (basal OXPHOS of CTRL 5.277 ± 0.402 vs. CB839 + T4 5.629 ± 0.634 pmolO 2 /min, ns, Fig. 3B), indicating that human cystic cells are dependent on glutamine entry in TCA. Interestingly, the combination of CB839 and T4 treatment boosted basal glycolysis more than T4 treatment alone (Fig. 3A), probably as a compensatory mechanism to sustain TCA using pyruvate instead of glutamine in the presence of CB839. 20 These findings confirm the glutamine dependency of human cystic cells observed in experimental models, 17 and further suggest that the anti-cystogenic effect of T4 is not predominantly mediated by regulating cellular energetics.
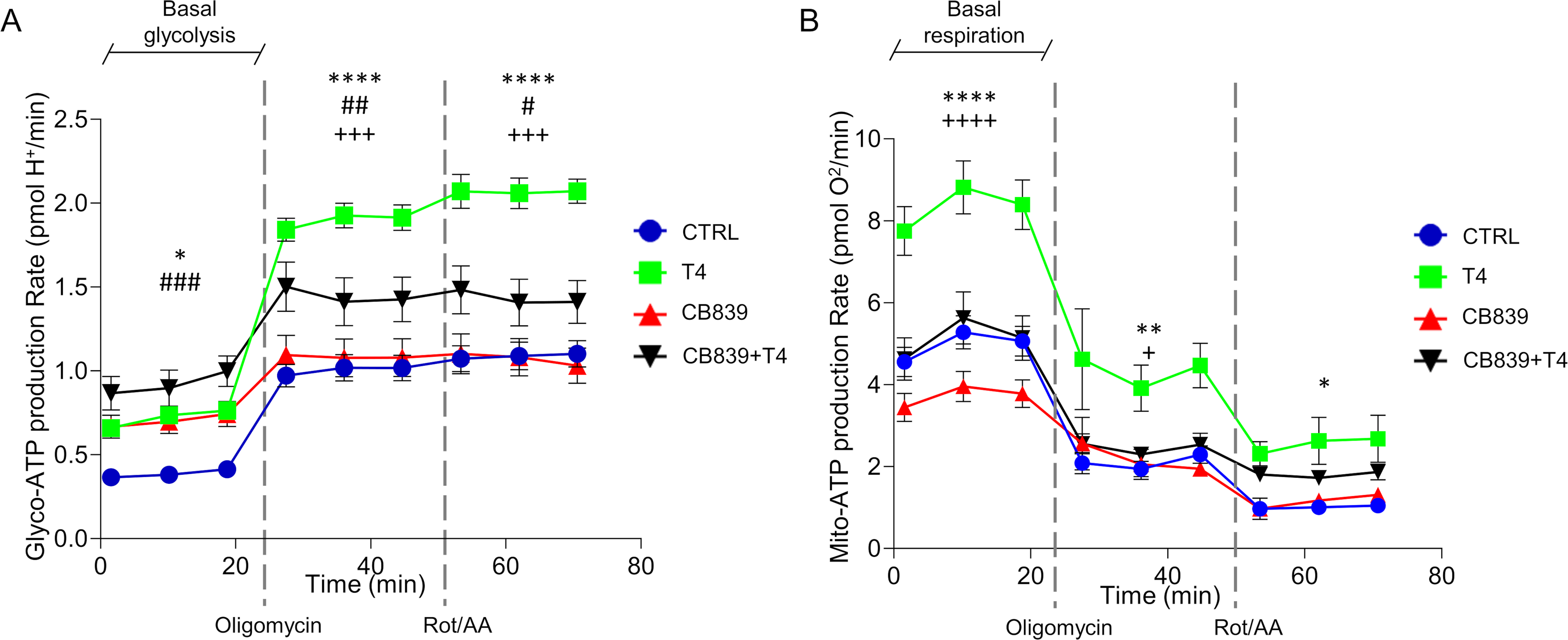
T4 treatment boosts metabolism in ADPKD cells.
T4 and ferroptosis
To determine whether T4 links metabolic rewiring to cell death pathways,18,37 we first performed targeted metabolomic profiling, which revealed an increase in multiple amino acids and metabolites following T4 treatment (Fig. 4A). Notably, levels of glutamine, cysteine, and glycine were remarkably elevated, suggesting enhanced activity of metabolic pathways controlling glutathione-dependent antioxidant defenses and ferroptosis susceptibility. 19
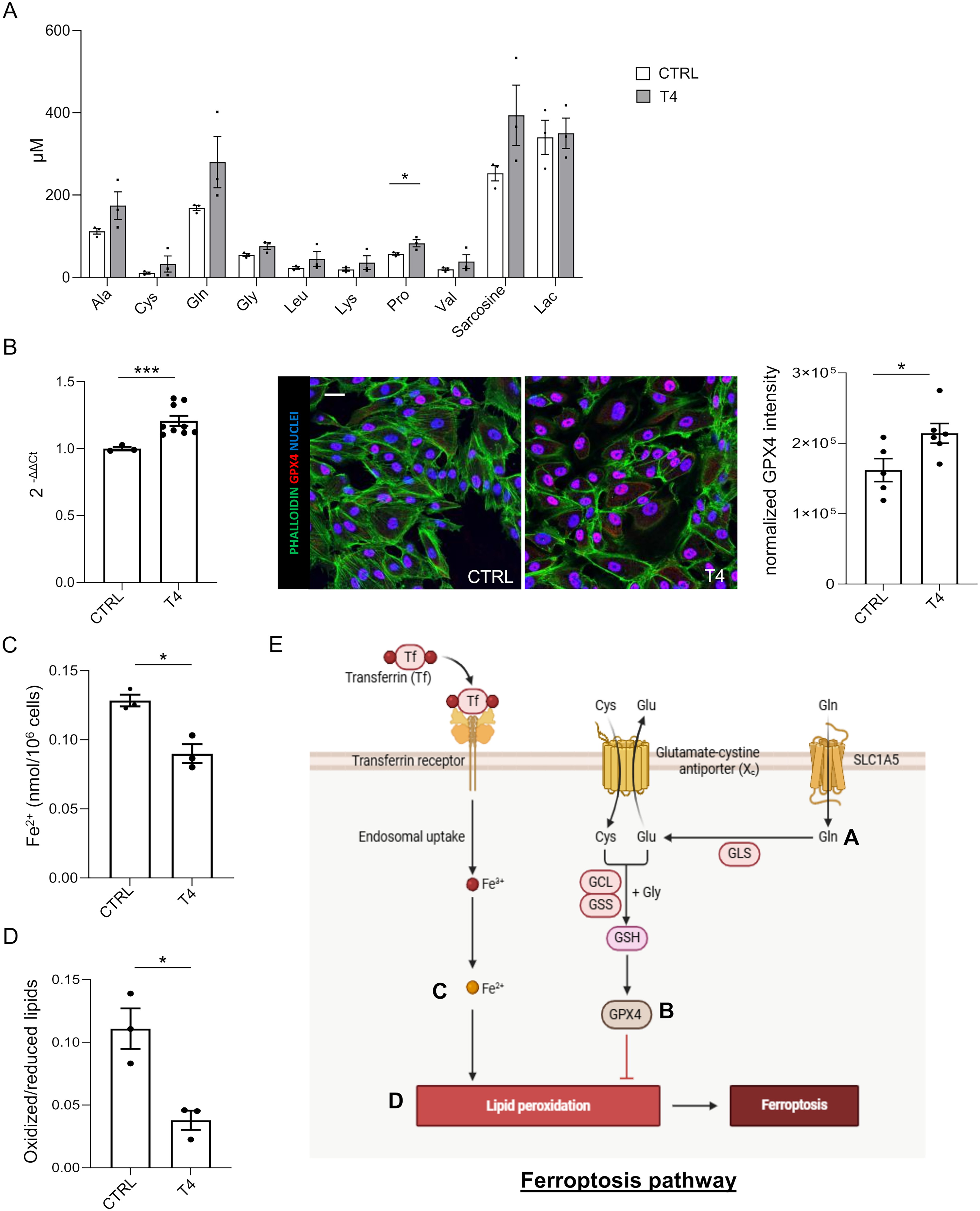
T4 treatment coordinately modulates metabolic and anti-oxidant pathways to inhibit ferroptosis.
Based on these findings, together with prior evidence implicating ferroptosis as a relevant cell death pathway in ADPKD,18,20 we focused on glutathione peroxidase 4 (GPX4), the only enzyme that can detoxify phospholipid hydroperoxides directly in membranes and prevent ferroptotic cell death.19,39 Consistently, qPCR analysis and immunofluorescence on ADPKD patient-derived cells (Fig. 4B) demonstrated a significant upregulation of GPX4 expression upon T4 treatment. Accordingly, intracellular Fe2+ levels (Fig. 4C) and lipid peroxidation (Fig. 4D), two key hallmarks of ferroptosis, were found reduced in ADPKD cells upon T4 treatment (CTRL 0.128 ± 0.004 vs. T4 0.090 ± 0.007 Fe2+ nmol/106 cells, p < 0.05, Fig. 4C; CTRL 0.111 ± 0.016 vs. T4 0.038 ± 0.008 Oxidized/reduced lipids, p < 0.05, Fig. 4D). Collectively, these results indicate that T4 coordinately suppresses ferroptosis in ADPKD cells (Fig. 4E).
T4 administration ameliorates renal structure and function in PCK rats
To assess the therapeutic potential of in vivo T4 administration, we used PCK rats, which share important pathophysiological features with human ADPKD.40,41 We observed that T4 supplementation significantly reduced the macrocystic area of PCK rats compared with untreated animals (Vehicle 9.255 ± 2.654 vs. T4 1.945 ± 0.850, p < 0.05, Fig. 5A). In addition, T4 administration decreased kidney weight—a marker of disease progression—in PCK rats (SD 0.981 ± 0.037 vs Vehicle 1.173 ± 0.025 p < 0.01; SD 0.981 ± 0.037 vs T4 1.091 ± 0.029 ns; Vehicle 1.173 ± 0.025 vs T4 1.091 ± 0.029 ns, Fig. 5B), although no effects were observed in kidney-to-body weight ratio (Fig. 5C). This may be due to the known metabolic effects of T4 on thermogenesis and calorie burn, 42 that led to weight loss in T4-treated animals (SD 240.2 ± 4.620 vs. Vehicle 271.3 ± 3.630 g, p = 0.0001; SD 240.2 ± 4.620 vs. T4 253.1 ± 3.535 g, ns; Vehicle 271.3 ± 3.630 vs. T4 253.1 ± 3.535 g, p < 0.001, Fig. 5D). Moreover, treating PCK rats with T4 effectively restored its serum levels to healthy controls (SD 3.682 ± 0.131 vs. Vehicle 3.257 ± 0.156 µg/mL, ns; SD 3.682 ± 0.131 vs. T4 4.256 ± 0.158 µg/mL, ns; Vehicle 3.257 ± 0.156 vs. T4 4.256 ± 0.158 µg/mL, p < 0.001 Fig. 5E), counteracting cyst growth and kidney function decline, as indicated by the inverse correlation between T4 levels and both the macrocystic area (Fig. 5F) and proteinuria (Fig. 5G).
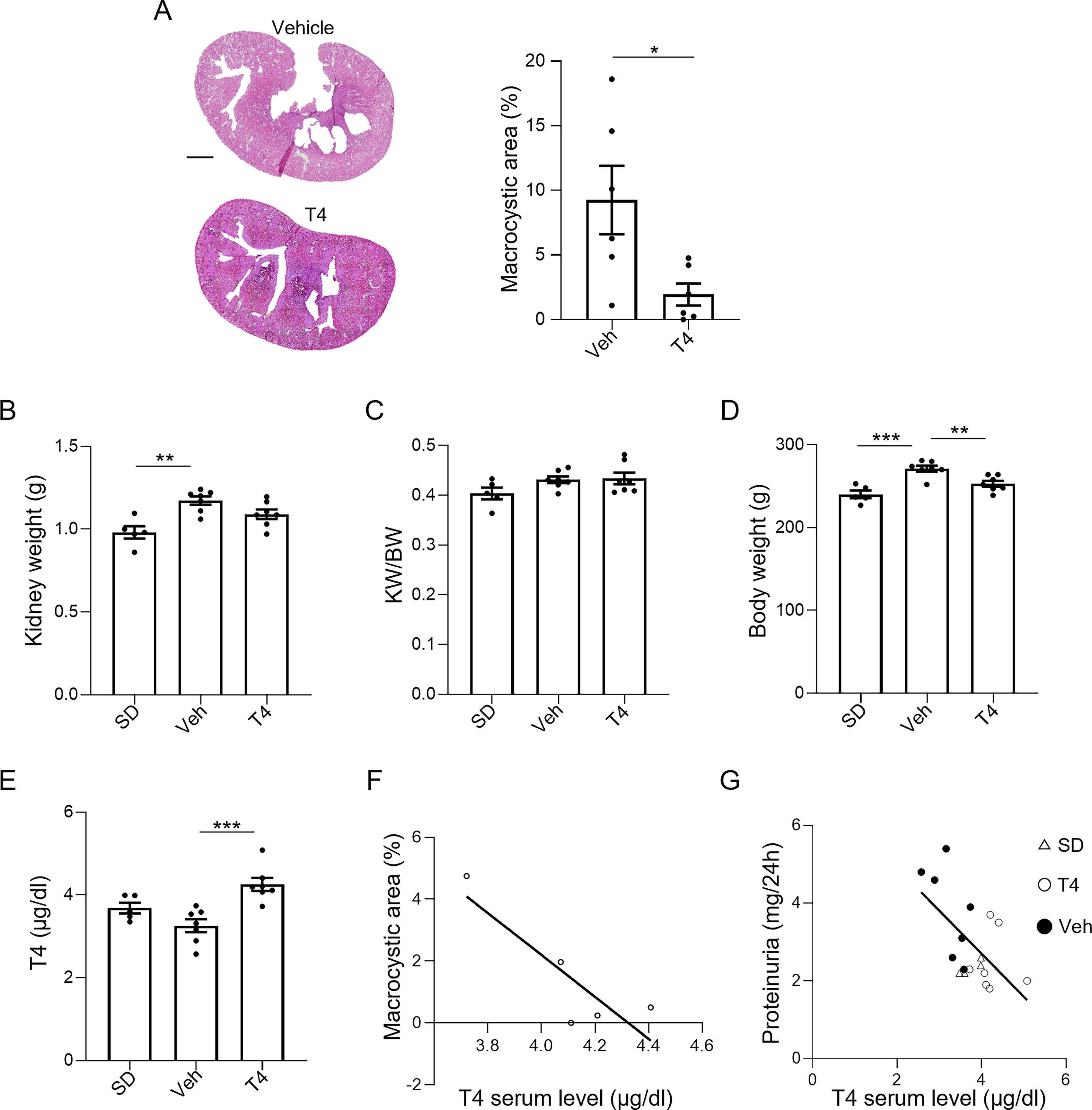
T4 administration restores T4 serum levels in PCK rats and improves kidney structure and function.
THs’ serum levels correlate with the severity of the disease
To assess whether THs levels are altered and whether these changes correlate with impaired renal function in ADPKD patients, we conducted the REORIENTED observational study. The analysis of THs serum levels showed that the mean values were in the physiological range, except for reverse T3 (rT3, a TH metabolite), which was found to be above the normal adult value (Table 1). When THs levels were correlated with the estimated glomerular filtration rate (eGFR) in the overall population of this study, a statistically significant inverse correlation was found between rT3 and eGFR values (R2 = 0.159, r = −0.397, p < 0.001) and a corresponding positive correlation was found between free T3 serum levels (fT3, the active form of TH) and eGFR values (R2 = 0.110, r = 0.332, p < 0.01, Fig. 6A). To investigate in greater depth whether changes in TH signaling are linked to impairments in kidney function, patients were divided into two groups that included subjects with a normal to mild (eGFR ≥ 60 mL/min/1.73 m2) or moderate to severe (eGFR < 60 mL/min/1.73 m2) decrease in renal function. Patients with moderate to severe impairment in renal function had significantly lower fT3 levels (<60 3.015 ± 0.037 vs. ≥ 60 3.190 ± 0.062 pg/mL, p < 0.05, Fig. 6B) and, concomitantly, a statistically significant increase in rT3 serum levels, compared with the other group (<60 462.0 ± 13.84 vs. ≥ 60 404.3 ± 12.54 pg/mL, p < 0.01, Fig. 6B). Most importantly, when TH serum levels and eGFR values were correlated in these two groups of patients, a significant negative correlation was found between rT3 and eGFR (R2 = 0.232, r = −0.482, p < 0.001) in subjects with a moderate to severe decrease in kidney function. Conversely, patients with normal to mild reduction in renal function exhibited a positive correlation between fT3 levels and eGFR values (R2 = 0.157, r = 0.397, p < 0.05, Fig. 6B).
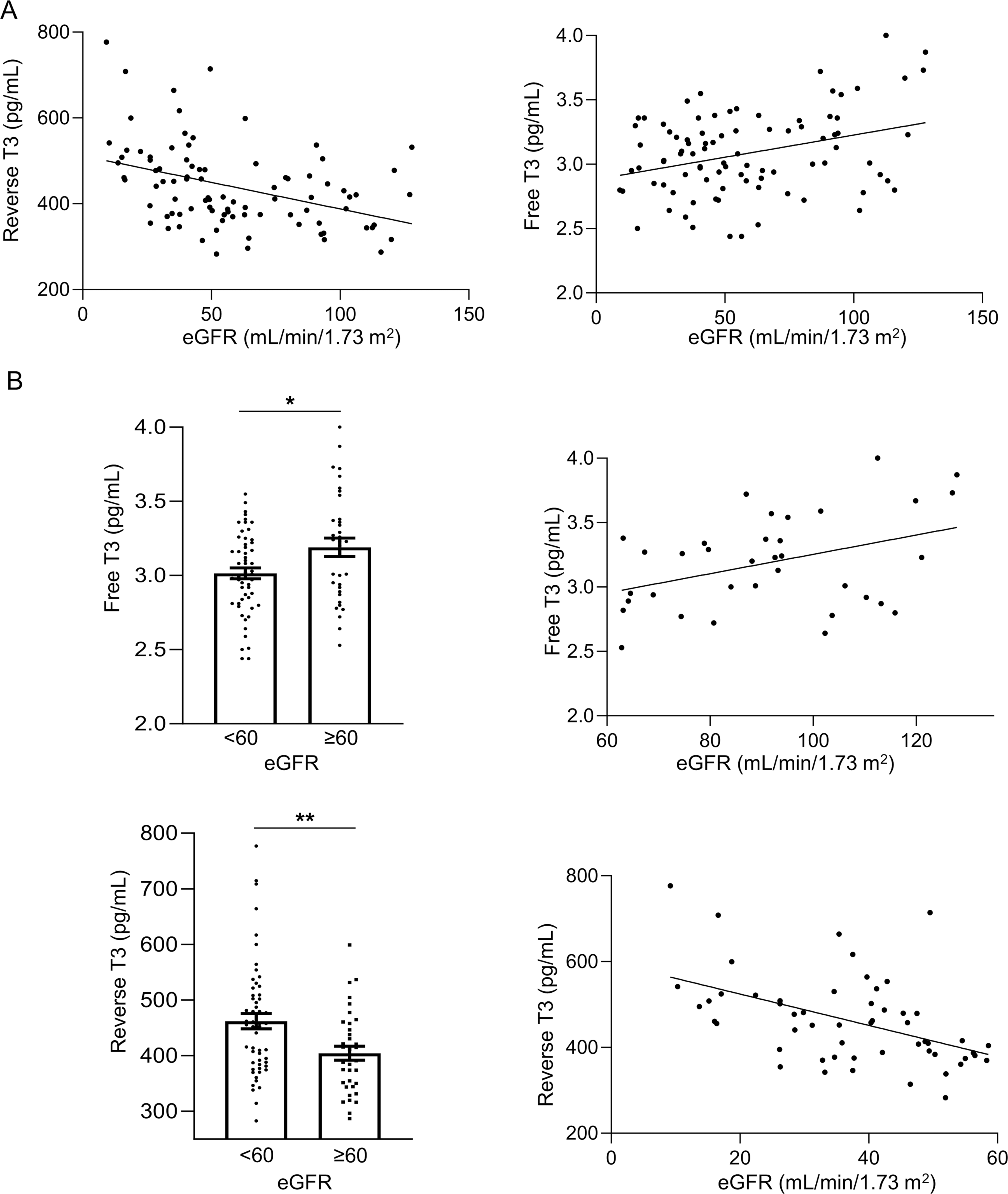
THs levels are altered in ADPKD patients and correlate with the progression of the disease.
Characteristics of Patients Enrolled in the Reoriented Study
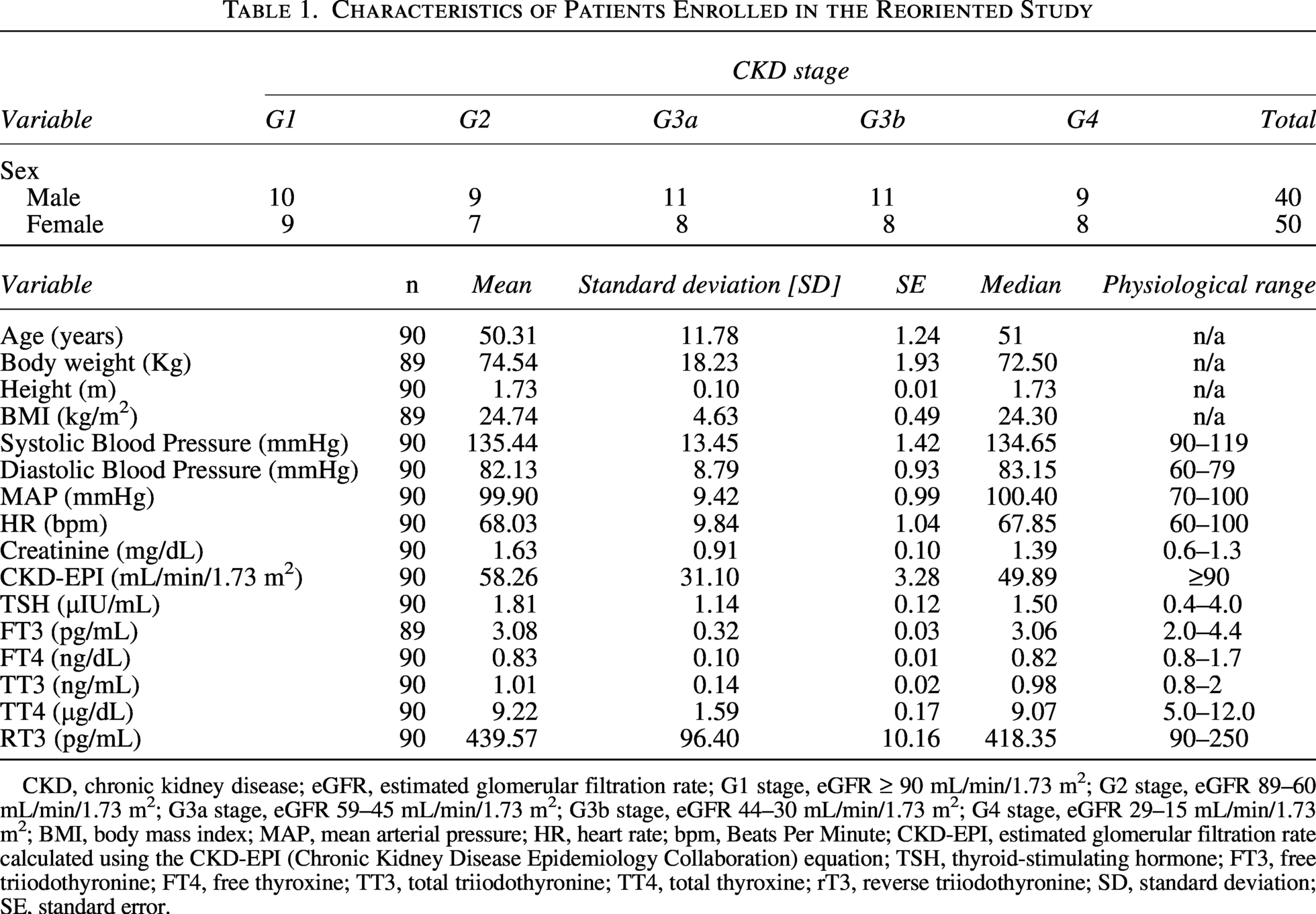
CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; G1 stage, eGFR ≥ 90 mL/min/1.73 m2; G2 stage, eGFR 89–60 mL/min/1.73 m2; G3a stage, eGFR 59–45 mL/min/1.73 m2; G3b stage, eGFR 44–30 mL/min/1.73 m2; G4 stage, eGFR 29–15 mL/min/1.73 m2; BMI, body mass index; MAP, mean arterial pressure; HR, heart rate; bpm, Beats Per Minute; CKD-EPI, estimated glomerular filtration rate calculated using the CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) equation; TSH, thyroid-stimulating hormone; FT3, free triiodothyronine; FT4, free thyroxine; TT3, total triiodothyronine; TT4, total thyroxine; rT3, reverse triiodothyronine; SD, standard deviation; SE, standard error.
Discussion
Our study demonstrates for the first time that THs, particularly T4, exert potent anti-cystogenic effects in ADPKD across both in vitro and in vivo preclinical models. Extending prior evidence that ERK activation drives cystogenesis and that its inhibition slows disease progression,43–45 we show that T4 suppresses cyst growth by downregulating ERK signaling via αvβ3 integrin, and by modulating key metabolic and anti-ferroptotic pathways.
Interestingly, while ERK inhibition is often linked to reduced glycolysis and glutamine metabolism, 46 our study revealed that T4’s anti-cystogenic effects do not stem from cellular energetics suppression. Instead, T4 promotes a selective metabolic enhancement, leading to the accumulation of key metabolites, including glutamine, cysteine, and glycine—precursors required for glutathione (GSH) biosynthesis and glutathione peroxidase 4 (GPX4) activity. These findings raise the possibility that T4 may strengthen the GSH–GPX4 anti-ferroptotic axis through increased availability of these metabolites.
We therefore examined ferroptosis—a form of regulated cell death controlled by GPX4 and driven by iron-dependent lipid peroxidation—which has been increasingly implicated in kidney disease.21,47,48 We found that T4 coordinately upregulates GPX4 and reduces lipid peroxidation and Fe2+ accumulation, identifying ferroptosis suppression as a key component of its protective effects. These results are in line with prior studies linking ferroptosis to cyst progression in ADPKD21 and describing anti-ferroptotic effects of THs in renal cells. 49 Interestingly, the T4-mediated inhibition of ERK1/2, a pathway known to modulate ferroptosis,50,51 is consistent with the observed increase in GPX4 expression and further supports the mechanistic link between T4 signaling and ferroptosis resistance.
Together, these data indicate that TH signaling exerts a multifaceted role in ADPKD pathobiology, with potential therapeutic implications through the coordinated regulation of proliferation, metabolism, redox balance, and cell survival.
Importantly, we validated these findings in vivo using PCK rats, an animal model of PKD, in which T4 treatment significantly reduced macrocystic area and improved overall renal structure and function. To assess the relevance of these findings in humans, we conducted the REORIENTED clinical study. Our analysis of TH serum levels revealed that, although most hormone values remained within the physiological range, reverse T3 (rT3) was consistently elevated. Crucially, rT3 levels negatively correlated with eGFR, while free T3 (fT3) exhibited a positive correlation. These relationships were particularly evident in patients with moderate to severe kidney dysfunction, which exhibit lower fT3 and higher rT3 levels than patients with normal to mild decrease in eGFR, possibly reflecting increased conversion of T4 in rT3 at the expense of the production of fT3.
These findings, along with our experimental data, suggest that alterations in TH signaling, particularly the shift toward the production of rT3, which is unable to carry out the biological activity of T4 and T3, may actively contribute to disease progression rather than simply reflecting declining renal function. Mechanistically, rT3 is known to compete with T3 for receptor binding, inhibiting its genomic and nongenomic actions,52,53 whereas elevated rT3 levels are linked to adverse outcomes in CKD and contribute to a state of functional hypothyroidism despite normal TSH levels.24,54 Therefore, rT3 emerges as a promising biomarker of disease severity and a potential indicator for a biomarker-guided approach that could enable personalized and controlled use of TH, only in patients who are most likely to benefit from it.
There are important limitations of this study that need to be acknowledged. First, while significant correlations between THs levels and disease severity were observed, longitudinal studies are needed to establish causality and determine whether TH alterations drive disease progression. Second, sex-specific responses to T4 treatment warrant further investigation, since our in vivo studies were conducted exclusively in female PCK rats. Third, a potential systemic T4 therapy carries risks and off-target actions—including cardiovascular side effects or the reduction in body weight observed in in vivo treatment—that may be particularly concerning for ADPKD patients who often present hypertension and cardiovascular comorbidities.
To mitigate potential complications associated with TH therapy, we are developing a nanomedicine-based delivery system 55 designed to selectively target renal cysts with T4, thereby maximizing therapeutic efficacy while minimizing systemic exposure.
In conclusion, our study demonstrates that THs play a central role in ADPKD pathobiology and holds promise as both a prognostic and therapeutic strategy. These findings pave the way for personalized treatment strategies aimed at slowing disease progression and potentially improving outcomes in patients with ADPKD. Further research is needed, however, to include more CKD patients as controls, refine dosing regimens, determine long-term efficacy, and evaluate potential synergy with existing therapies.
Authors’ Contributions
A.M.L. designed and performed the REORIENTED clinical study, collected, analyzed, interpreted the data and wrote the article; L.L. performed in vitro experiments, analyzed and interpreted the data, prepared the figures and wrote the article; M.T. conceived, designed and performed the REORIENTED clinical study; A.D.V. performed in vitro experiments; Domenico C. performed animal studies; A.V. conceived and designed the REORIENTED clinical study; T.P. performed the statistical analyses for the REORIENTED clinical study, V.B. carried out in vitro experiments; S.B. carried out in vitro experiments. Daniela C. performed animal studies; L.B. performed and interpreted metabolomic studies; C.G. performed the REORIENTED clinical study; N.R. conceived and designed the REORIENTED clinical study; G.R. critically revised the article and approved the final version of the article; C.X. conceived, designed and supervised the study and the REORIENTED clinical trial, analyzed and interpreted data, wrote the article. All authors reviewed the final version of the article and took responsibility for the content.
Footnotes
Acknowledgments
The authors are grateful to Ariela Benigni and Susanna Tomasoni for their fruitful comments on the article, Kerstin Mierke for the excellent editing work on the article and to Benedetta Petracca, Melissa Balsamo and Francesca Ricci for their technical contributions to the experiments. The authors thank all the patients who participated in this REORIENTED study for their valuable contribution.
Data Availability Statement
Author Disclosure Statement
Authors declare no competing interests.
Funding Information
A.M.L. was recipient of fellowship from Fondazione Aiuti per la Ricerca sulle Malattie Rare (A.R.M.R.), Bergamo (Italy). L.L. is recipient of fellowship from Fondazione Aiuti per la Ricerca sulle Malattie Rare (A.R.M.R.), Bergamo (Italy). Domenico C. was recipient of fellowship from Fondazione Aiuti per la Ricerca sulle Malattie Rare (A.R.M.R.), Bergamo (Italy). Valerio Brizi was recipient of fellowship from Fondazione Aiuti per la Ricerca sulle Malattie Rare (A.R.M.R.), Bergamo (Italy). M.T., A.D.V., A.V., T.P., S.B., Daniela C., L.B., C.G., N.R., G.R., and C.X.: No funding information to declare.
This research was funded by Fondazione Telethon ETS (project number: GMR23T2014) and Fondazione Terzi-Albini.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.