
Abstract
Objective
Facial dysostosis is a group of rare craniofacial congenital disabilities requiring multidisciplinary long-term care. This report presents the phenotypic and genotypic information from South India.
Design
The study is a case series.
Setting
This was an international collaborative study involving a tertiary craniofacial clinic and medical genetics unit.
Patients, Participants
The participants were 9 families with 17 affected individuals of facial dysostosis.
Intervention
Exome analysis focused on known genes associated with acrofacial and mandibulofacial syndromes.
Main Outcome Measure
The outcome measure was to report phenotyptic and genetic heterogeneity in affected individuals.
Results
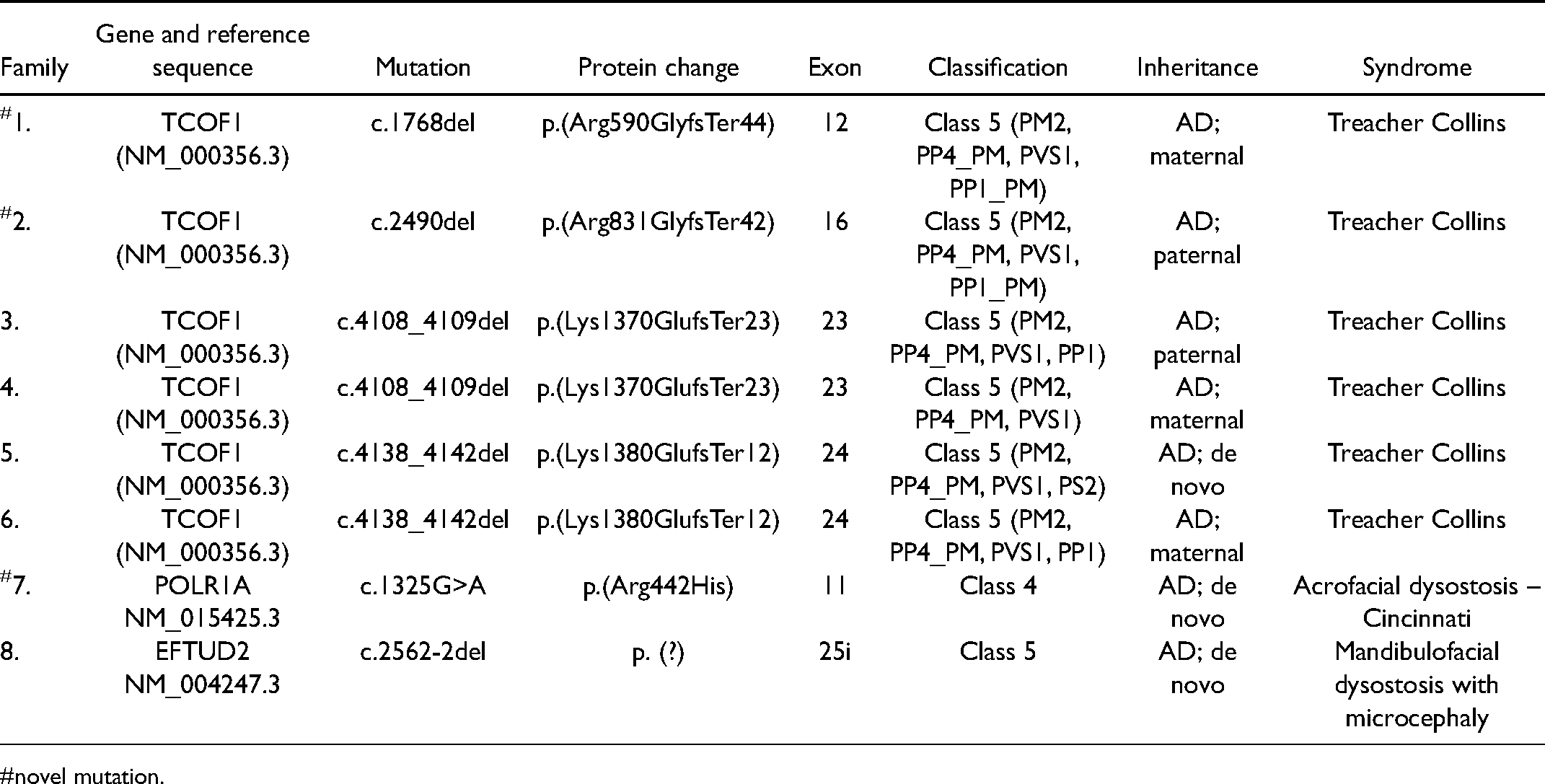
A Tessier cleft was seen in 7 (41%), lower eyelid coloboma in 12 (65%), ear anomalies in 10 (59%), uniolateral or bilateral aural atresia in 4 (24%), and deafness in 6 (35%). The facial gestalt of Treacher Collins syndrome (TCS) showed extensive phenotypic variations. Pathogenic variants in TCOF1 (Treacher Collins syndrome) were seen in six families, POLR1A (acrofacial dysostosis, Cincinnati type) and EFTUD2 (mandibulofacial dysostosis with microcephaly) in one each. One family (11.1%) had no detectable variation. Five out of six probands with Treacher Collins syndrome had other affected family members (83.3%), including a non-penetrant mother, identified after sequencing.
Conclusion
Our report illustrates the molecular heterogeneity of mandibulofacial dysostosis in India.
Keywords
Introduction
The face develops between the fourth and eighth week of gestation. A critical step in morphogenesis is the formation, migration, and differentiation of neural crest cells in the facial region (Trainor, 2010). The interaction between the three primary embryonic layers of first and second pharyngeal arches, the neural crest cells, the mesenchyme, and several signaling pathways are complex, making the craniofacium susceptible to developmental defects.
Facial dysostoses are autosomal disorders with bilateral symmetrical hypoplasia of the maxilla and zygoma. Processes affecting the formation and migration of the neural crest cells due to defective ribosome biogenesis and ribonucleoprotein metabolism result in classical phenotypes. These syndromes are grouped into mandibulofacial dysostosis (MFD) or acrofacial dysostosis (AFD), depending on the absence or presence of additional limb defects (Terrazas et al., 2017). Children with these disorders can have life-threatening upper airway obstruction at birth and require to undergo multi-stage surgeries late into adulthood for good functional and cosmetic outcome with devastating emotional and psychological issues (Smith et al., 2021).
Treacher Collins syndrome (TCS; Online Mendelian Inheritance in Man (OMIM)#154500) is the most common MFD and is considered to have an incidence of 1 in 50 000 live births (Hennekam et al., 2010). The causative genes are TCOF1, POLR1C or POLR1D involved in ribosome biogenesis. Variants in TCOF1 account for 80% of all TCS cases (Vincent et al., 2016). The AFD, Cincinnati type (AFDCIN; OMIM#616462), results from defective ribosomal RNA transcription due to loss of function variations in the POLR1A gene (Weaver et al., 2015; Terrazas et al., 2017). Variations in the SF3B4 gene and EFTUD2 gene cause defective pre-messenger RNA splicing and Nager syndrome (OMIM#154400) and MFD with microcephaly (MFDM; MFD Guion-Almeida type; OMIM#610536), respectively (Lehalle et al., 2015; Terrazas et al., 2017). The DHODH gene is linked to the Miller syndrome (OMIM#263750), but the exact mechanism causing the facial dysostosis is unclear. The true incidence of other facial dysostoses is not available.
MFDs are well described in Caucasians, but the number of reports among Asians is limited (Horiuchi et al., 2005; Chen et al., 2018; Yu et al., 2018; Kantaputra et al., 2020). This report presents the phenotypic and genotypic information of 9 families with facial dysostosis from South India, specifically the states of Kerala and Karnataka.
Materials and Methods
We evaluated 9 families with facial dysostosis visiting a craniofacial clinic in a tertiary teaching hospital in South India. Pedigrees and phenotypes were documented. Consents and assents (where applicable) were obtained for reporting clinical photographs, clinical data, and genetic testing results. The Institutional Ethical Committee for Human Research approved the study (No. INST.EC/2017-18/005).
For exome sequencing, genomic DNA was enriched with the Sureselect All Exon v7 kit (Agilent Technologies) followed by sequencing on a NovaSeq 6000 platform (Illumina). After removal of duplicate reads, variants were annotated using in-house (Seqplorer) software based on commercially available software (CLC Genomics). Mean coverage was >80 reads. Initial analyses focused on known genes associated with acrofacial and mandibulofacial phenotypes. Regions with a coverage <20 reads were additionally analyzed with Sanger sequencing. Variant prioritization was based on segregation and in silico prediction (pLi for premature truncating mutations, Z-score and rare exome variant ensemble learner (REVEL) score for missense mutations and Human splice finder/MaxENTscan score for splice site mutations).
The variants are presented according to the Human Genome Variation Society and American College of Medical Genetics and Genomics nomenclature. The potential pathogenicity of these variants was confirmed using standard genome databases.
Descriptive data is presented. The categorical data is presented as percentages.
Results
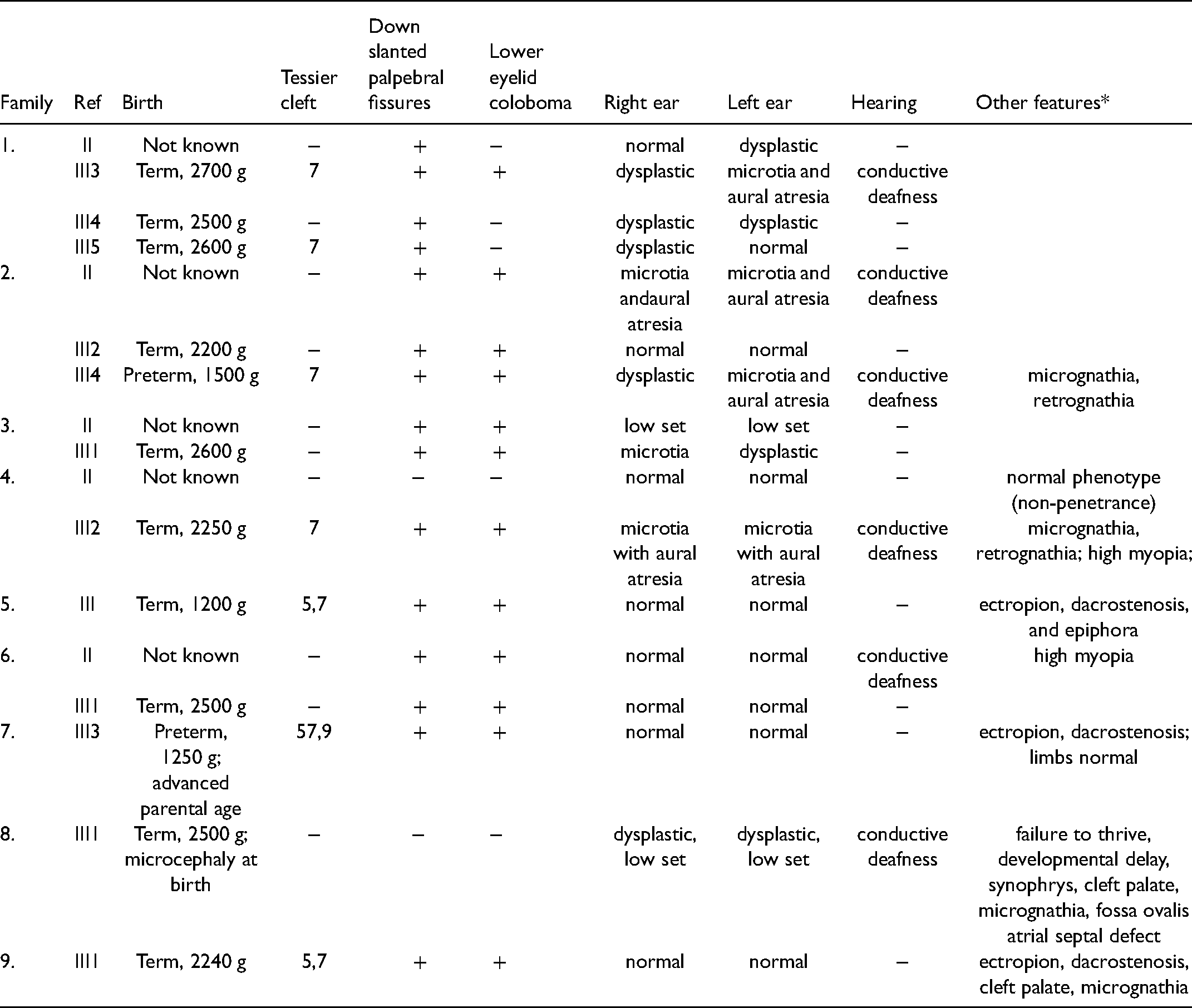
We identified 17 individuals from 9 families with an MFD. The pedigrees, demographic, and clinical findings are presented in Table 1, Supplementary Figure S1, Figures 1–3. A Tessier cleft was seen in 7 (41.1%), lower eyelid coloboma in 12 (64.7%), ear anomalies in 10 (58.8%), unilateral or bilateral aural atresia in 4 (23.5%), deafness in 6 (35.3%), and cleft palate in 2 (11.2%). The facial gestalt of TCS showed intrafamilial and interfamilial variations (Figure 1). This and the other images in the report are representative of the facial features seen in these individuals.

The facial features of 12 of the affected members with Treacher Collins syndrome (TCS) of families 1 to 6. The facial gestalt illustrates the phenotypic heterogeneity. The figure includes phenotype of the nonpenetrant mother of family 4 (bottom panel). Two other affected members of family 2 are not shown.
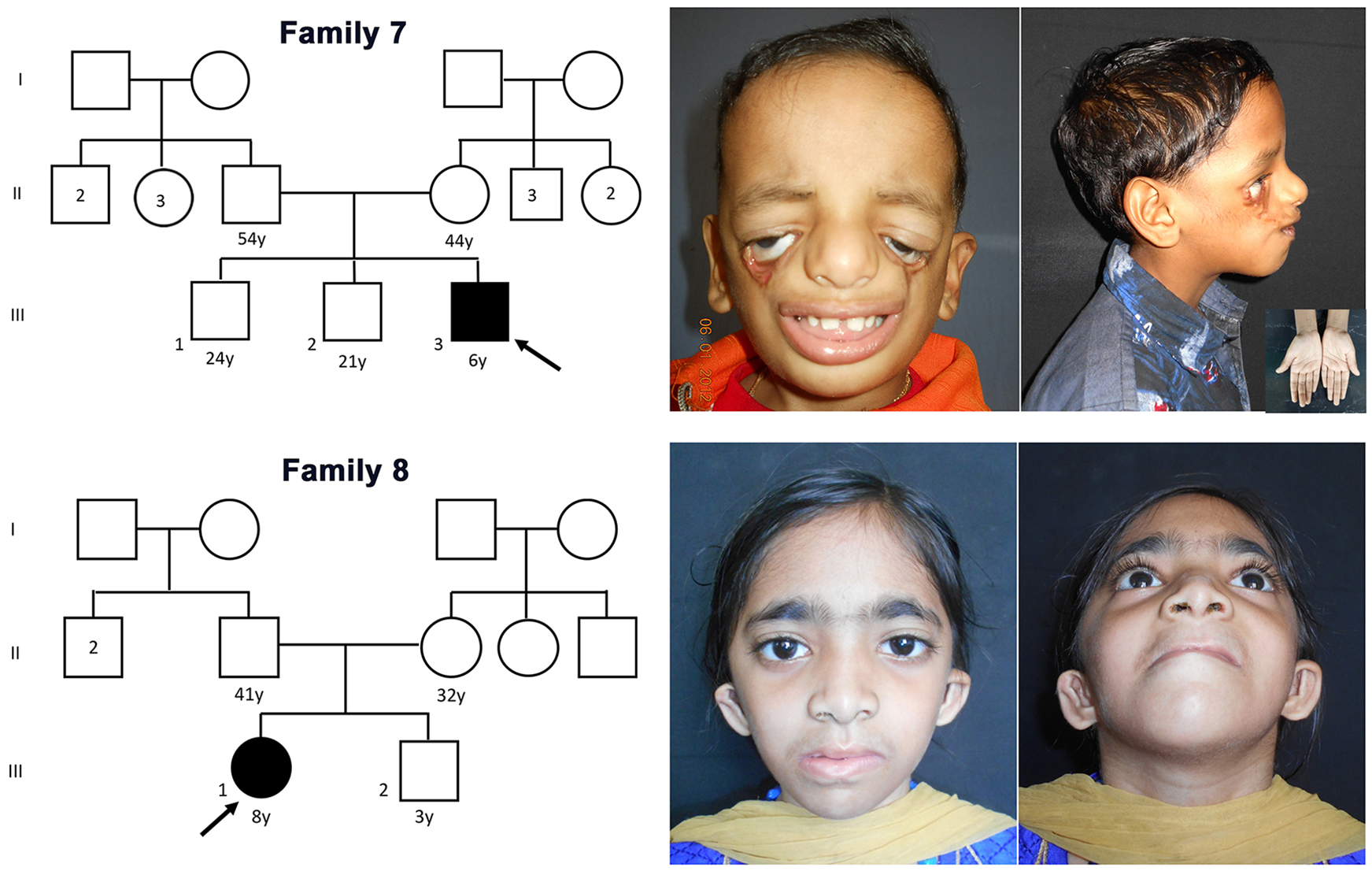
The pedigrees of families 7 and 8 along with the facial features of the probands. Note the severe malar hypoplasia with Tessier cleft and normal limbs (inset) in the proband of family 7 (top panel). The lower panel shows the proband of family 8 with features of mandibulofacial dysostosis with microcephaly.
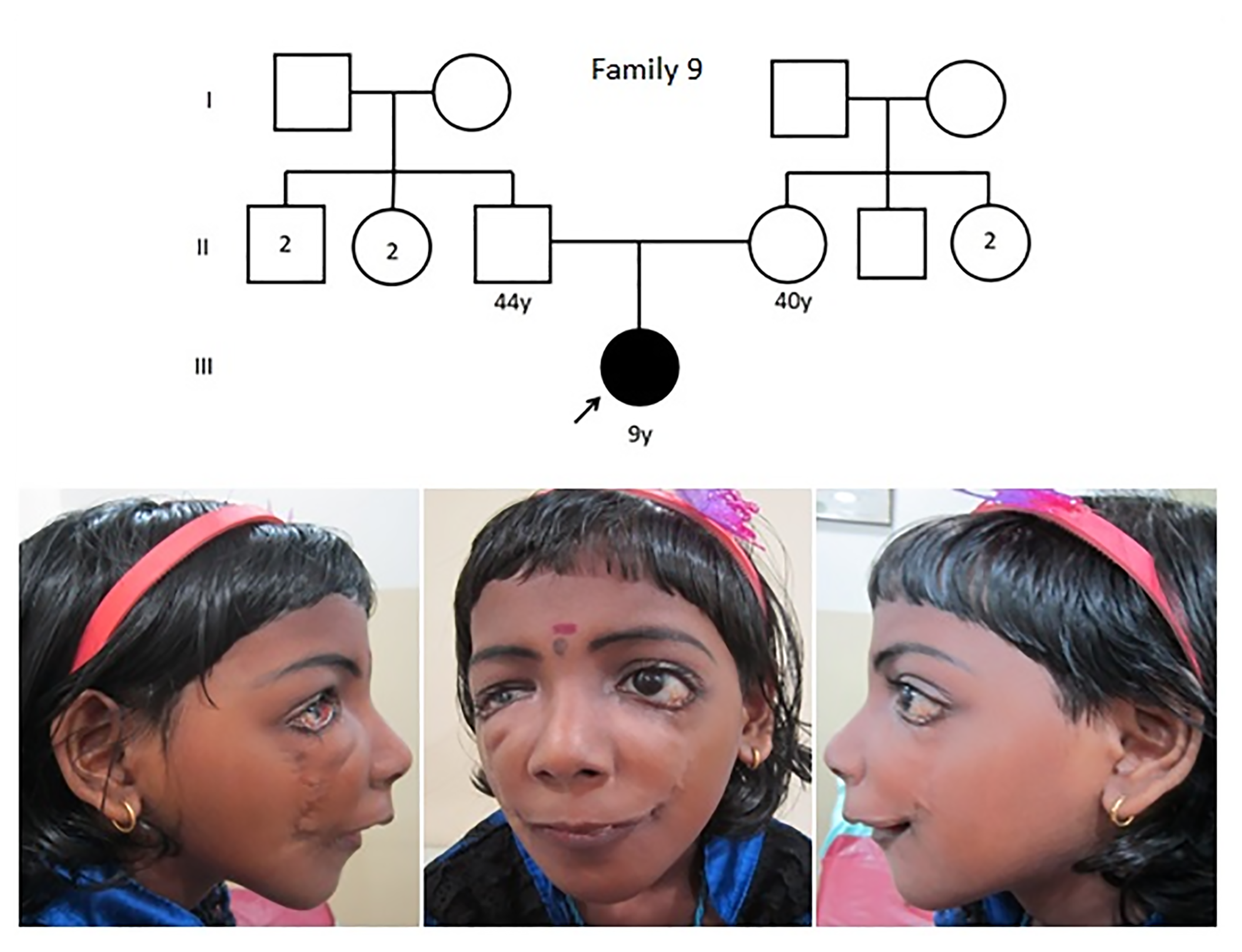
The pedigree of family 9 with proband showing the facial gestalt of mandibulofacial dysostosis. Sequencing was negative for a variation.
Phenotypic Presentation of all Affected Individuals.
Children with MFD require sequential individualized surgical correction to preserve vital functions and achieve good cosmesis (Supplementary Figure S2). In a severe phenotype with life-threatening upper airway obstruction, tracheostomy or glossopexy and repair of choanal atresia is required in the newborn period. If it persists, mandibular advancement with distraction osteogenesis is performed. The Tessier 7 cleft is corrected at 24 months of age along with release of glossopexy and correction of middle ear atresia so that speech and hearing rehabilitation can begin. Incremental expansion of the skin overlying the zygoma is done between 5 and 8 years of age along with correction of eyelid colobomata, so that definitive implants can be inserted by 14 years. Orthodontic treatment of the maxilla and the mandible is initiated at 8 years to achieve a stable occlusion that will aid in function and future cosmetic repair. Reconstruction of the external pinna is also carried out at 8 years of age. Definitive cosmetic repair of the maxillo-mandibular skeleton is done at 14 years of age and a rhinoplasty, if needed, after 15 years. These surgeries also help improve occlusion and upper airway issues. All through these procedures, great anesthetic challenges are faced by the patient and the surgical team.
Exome sequencing identified a pathogenic variant in 8 (88.9%) families (Table 2). Families 1 to 6 harbored pathogenic class 5, frameshift variants in TCOF1, confirming TCS (Figure 4). None of the individuals with TCS had extrafacial manifestations. Five of six probands with TCS had other affected family members (83.3%), including a non-penetrant mother in family 4 (Figure 1), identified after sequencing. We noted two novel variants involving exon 12 (family 1) and exon 16 (family 2). Two variants involving exons 23 and 24 were seen in more than one family.
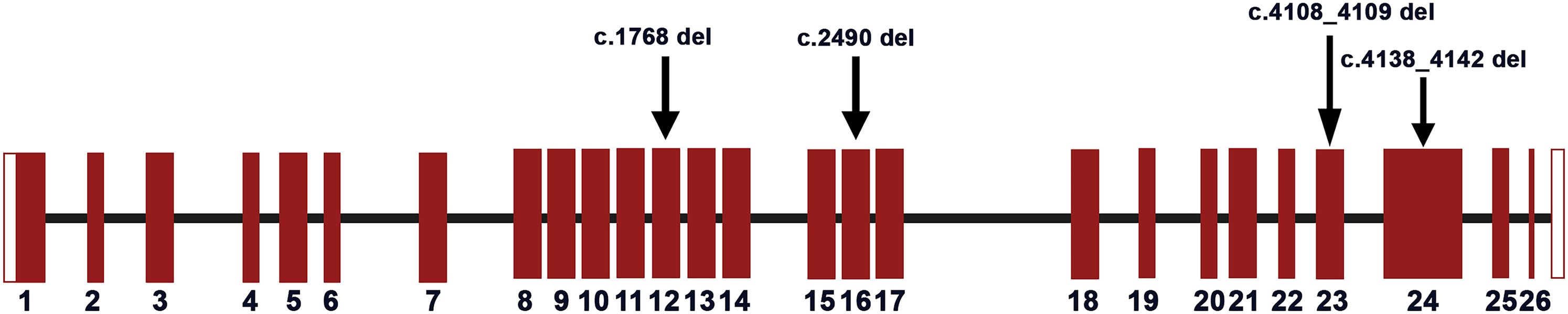
A schematic representation of the TCOF1 gene with the distribution of the causative variants identified in Indian patients in this study. The vertical bars represent the coding exons and the connecting lines between the bars represent the introns, represented as per the graphical summary of TCOF1 transcript in Ensembl (RefSeq: NM_001371623). The mutations identified in the probands are marked with arrows.
Molecular Findings.
novel mutation.
The proband of family 7 had a de novo pathogenic variant in POLR1A indicating AFDC. He had severe malar hypoplasia with Tessier cleft, but normal limbs (Figure 2 top panel). Parental age was advanced. The proband of family 8 with the typical craniofacial features (Figure 2 bottom panel) of MFDM showed a de novo pathogenic variant in EFTUD2. She had prenatal-onset microcephaly with a birth occipito-frontal circumference (OFC) of 29.0 cm (−3.9SD). She required surgeries for atrial septal defect and cleft palate in infancy. She had growth failure and global developmental delay. At 8 years of age, her OFC was 45.0 cm (−5.4SD), suggesting progressive microcephaly. Bone age was normal.
The proband of family 9 did not show a pathogenic variant in known genes for MFD/AFD, despite a typical phenotype (Figure 3).
Discussion
The study illustrates the variable expression and molecular heterogeneity in Indian patients with facial dysostoses, underscoring the importance of molecular evaluation in MFD. The causal pathogenic variant detection rate using exome sequencing was close to 90% in our study, which is slightly <96% reported by Vincent et al. (2016).
The phenotypic heterogeneity and intra- and interfamilial variable expression, previously well documented (Vincent et al., 2016; Terrazas et al., 2017), was seen in our patients as well. However, we found higher rates (>80%) of a positive family history compared to 17% to 42% in various case series (Splendore et al., 2000; Teber et al., 2004; Vincent et al., 2016; Chen et al., 2018). Parents in our study generally had milder phenotypes and normal growth and intellect. This might relate to the fact that prenatal diagnosis is not widely available, and medical termination of pregnancy is culturally and religiously not acceptable in India.
The TCOF1 pathogenic variants reported are suspected to cause haploinsufficiency. We identified two novel variants c.1768del (exon 12) and c.2490del (exon 16). The pathogenic variants in exon 23 (c.4108_4109del) and exon 24 (c.4138_4142del) have been previously reported (Ellis et al., 2002; Kantaputra et al., 2020), and are recurrent in our cohort. Pathogenic variants in exons 23 and 24 are common in TCS (Vincent et al., 2016; Chen et al., 2018).
AFDCIN is a recently described syndrome. The proband harboring a c.1325G>A in POLR1A had severe facial dysmorphisms with no limb anomalies, similar to observations in two of the three cases described by Weaver et al. (2015). All the three children reported by Weaver et al. (2015) also had unique de novo variations. The pathogenic variant we observed has not been reported previously and is not present among the 34 pathogenic variants reported in the Leiden Open Variation Database (LOVD) (https://databases.lovd.nl/shared/refseq/POLR1A_NM_015425.3_table.html).
Progressive microcephaly during the first year of life and intellectual disability are features of MFDM. Cleft palate is seen in about 40% to 45% and congenital heart defect in 32% to 35% (Lehalle et al., 2015; Abell et al., 2021). Splice site variants (including deep intronic variants) account for 43% of MFDM cases and may be missed by exome and or Sanger sequencing of genomic DNA. The variant c.2562-2del we identified in the EFTUD2 gene is previously reported by Lehalle et al. (2015) and this variant is predicted to disrupt the highly conserved acceptor splice site of exon 26.
In conclusion, we illustrate the molecular heterogeneity of MFD in an Indian cohort. We found a higher proportion of familial TCS, and lower variation detection rates, likely reflecting different prenatal practices and suggesting further molecular heterogeneity.
Supplemental Material
sj-jpg-1-cpc-10.1177_10556656211050006 - Supplemental material for Phenotypic and Molecular Heterogeneity in Mandibulofacial Dysostoses: A Case Series From India
Supplemental material, sj-jpg-1-cpc-10.1177_10556656211050006 for Phenotypic and Molecular Heterogeneity in Mandibulofacial Dysostoses: A Case Series From India by Rathika D. Shenoy, Vikram Shetty, Annelies Dheedene, Björn Menten, Dechamma Pandyanda Nanjappa, Gunimala Chakraborty, Patrick Sips, Anne de Paepe, Bert Callewaert and Anirban Chakraborty in The Cleft Palate-Craniofacial Journal
Supplemental Material
sj-jpg-2-cpc-10.1177_10556656211050006 - Supplemental material for Phenotypic and Molecular Heterogeneity in Mandibulofacial Dysostoses: A Case Series From India
Supplemental material, sj-jpg-2-cpc-10.1177_10556656211050006 for Phenotypic and Molecular Heterogeneity in Mandibulofacial Dysostoses: A Case Series From India by Rathika D. Shenoy, Vikram Shetty, Annelies Dheedene, Björn Menten, Dechamma Pandyanda Nanjappa, Gunimala Chakraborty, Patrick Sips, Anne de Paepe, Bert Callewaert and Anirban Chakraborty in The Cleft Palate-Craniofacial Journal
Footnotes
Acknowledgements
This project is a part of the collaborative research activities between Ghent University, Ghent, Belgium and Nitte (Deemed to be University), Mangalore, India under the Indo-Belgian Research and Technology Cooperation Topping-up Grant.
Data availability
Data will be available on reasonable request from the corresponding author.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by The Special Research Fund of Ghent University, Belgium (grant number BOF16/STA/045, NUFR2/2018/10/30, and V.25011/380/2015-GIA/HR), and also a Topping-up Grant, awarded to AC and AP by Department of Science & Technology, Government of India (INT/BELG/P-08/2017) and BELSPO, Belgium (BL/02/IN13), respectively. The financial support from Nitte (Deemed to be University) in the form of an intramural research grant (NUFR2/2018/10/30) awarded to AC and RS is gratefully acknowledged.
Ethical considerations
The Institutional Ethics Committee, Nitte University Centre for Science Education & Research, Mangalore, India, approved the study (INST.EC/2017-18/005). The study conformed to recognized standards as per the WMA declaration of Helsinki.
Data availability
Data will be available on reasonable request from the corresponding author.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.