
Abstract
Nager syndrome (NS) is a rare disease marked with craniofacial and preaxial limb anomalies. In this report, we summarized the current evidence to determine a possible genotype–phenotype association among NS individuals. Twenty-four articles comprising of 84 NS (including 9 patients with a severe form of NS [Rodriguez syndrome]) patients were examined, of which 76% were caused by variants in SF3B4 (OMIM *605593, Splicing Factor 3B, Subunit 4). Within the SF3B4 gene, variants located in exon 3 commonly occurred (20%) from a total identified variant, while hotspot location was identified in exon 1 (12%), and primarily occurred as frameshift variants (64%). Thirty-five distinct pathogenic variants within SF3B4 gene were identified with two common sites, c.1A > G and c.1060dupC in exons 1 and 5, respectively. Although no significant genotype–phenotype association was found, it is notable that patients with frameshift SF3B4 variants and predicted to lead to nonsense-mediated RNA decay (NMD) of the transcripts tended to have a more severe clinical manifestation. Additionally, patients harboring variants in exons 2 and 3 displayed a higher proportion of cardiac malformations. Taken together, this article summarizes the pathogenic variants observed in SF3B4 and provides a possible genotype–phenotype relationship in this disease.
Introduction
Nager syndrome (NS, preaxial acrofacial dysostosis or acrofacial dysostosis type 1 [AFD1]; MIM #154400) is a rare genetic disease that was first described in 1948 by Nager and de Reyenier, and is characterized by craniofacial anomalies and limb malformations.1–3 To date, less than 100 NS cases have been reported. 1 The identification is often challenging due to overlapping phenotypes with other craniofacial anomalies such as Miller syndrome, mandibulofacial dysostosis with microcephaly (MFDM), and Treacher Collins syndrome (TCS). Recently, however, the lowered cost of sequencing has allowed physicians to identify the syndrome by genetic testing.
Although most cases are the result of de novo variants, evidence suggests that NS could be inherited both in autosomal dominant and recessive manner.4–6 Bernier et al. first described that haploinsufficiency of SF3B4 is responsible for more than 50% of clinically diagnosed patients, which was also supported by the study of Petit et al. and Czeschik et al. in the following years.3–5 However, it should be noted that no clear evidence of genotype–phenotype association has been reported in NS. 2 This is possibly due to the high variability of clinical expression among NS individuals and the limited reported cases. This report summarizes the current evidence and tries to seek for the possible genotype–phenotype correlations in NS.
Materials and Methods
Literature Search and Data Extraction
Dataset of the current analysis was identified by querying PubMed and Scopus with search terms such as “Nager syndrome”, “preaxial acrofacial dysostosis”, “SF3B4”, and “clinical features” up to June 12, 2021. Using this query, 122 and 41 records were retrieved from PubMed and Scopus, respectively (Figure 1). Records included in the study are (1) describing both completed molecular genetic testing and NS clinical features and (2) evaluating severe manifestation of NS (Rodriguez syndrome). 7 Reference lists of retrieved articles were also manually screened to identify additional or potentially missing eligible studies. Data were retrieved from all selected publications and extracted as follows: (1) name of the first author; (2) year of publication; (3) country of origin; (4) age; (5) genetic information (including molecular genetic anomaly, cDNA position, exon location, and inheritance); and (6) 18 clinical features of NS (see Supplementary Table 1). In this study, all of the included SF3B4 variants were pathogenic or likely pathogenic (only study from Chummun et al. 8 was not determined due to lack of genetic information, Supplementary Table 1). All other variants of interests were not included in the analysis.
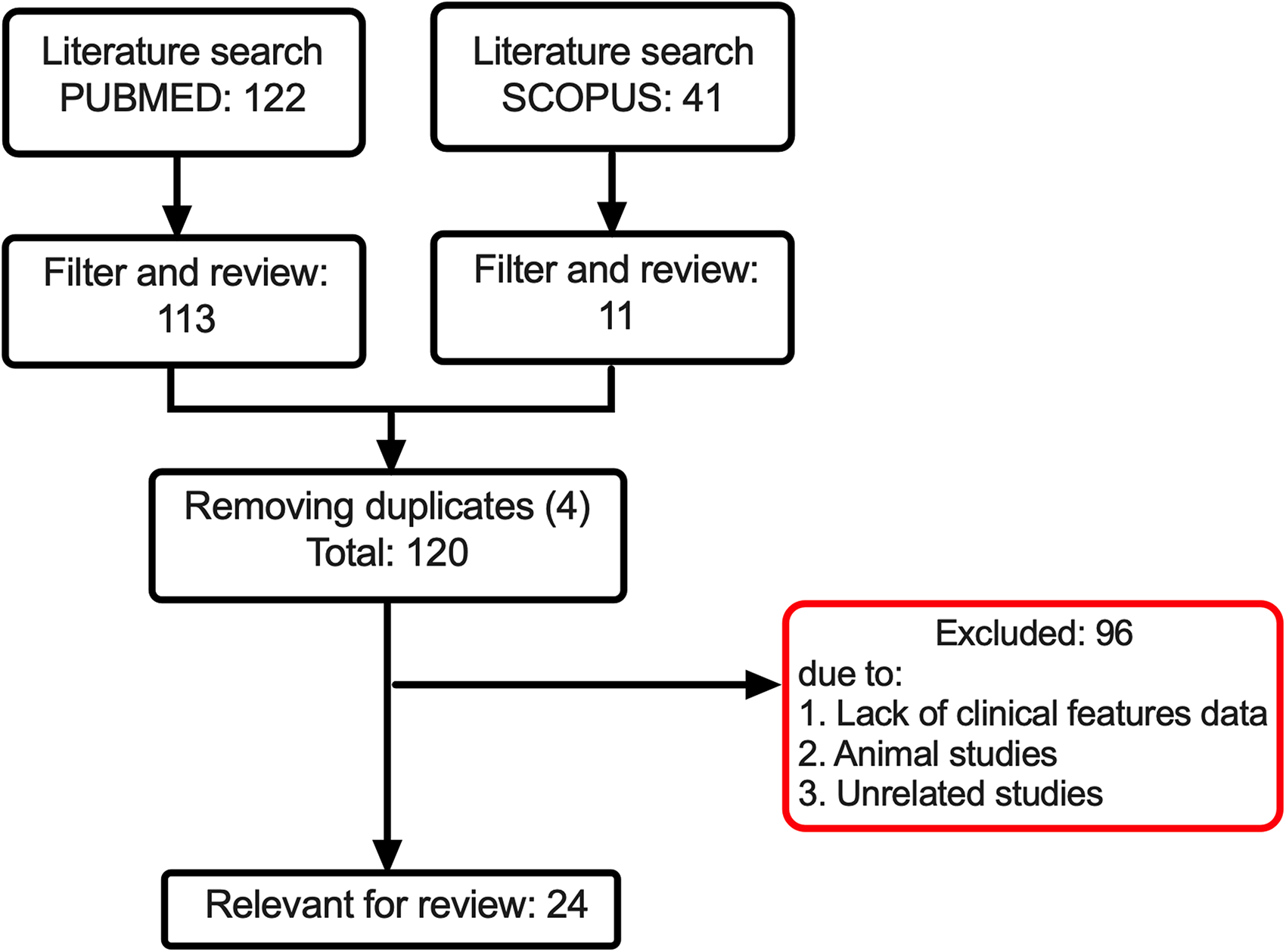
Study flowchart for literature search and selection.
Statistical Analysis
The chi-square test was used to compare the proportions of qualitative variables between two groups (classical Fisher’s exact test was conducted if more than 20% of the expected frequencies are less than 5). If the number of the group exceeds 2, the chi-square test (alternatively, if more than 20% of the expected frequencies are less than 5 were observed, the Fisher-Freeman-Halton exact test was applied) followed by pairwise comparisons and Bonferroni correction was performed. All tests were two-sided with the significance level at .05. Statistical analysis was performed using the SPSS Version 25.
Results
Study Population and Hotspot Mutation in SF3B4
A total of 120 articles were screened, among which 24 passed the criteria and were further evaluated1–24 (Figure 1). Details of the retrieved studies are shown in Supplementary Table 1. Eighty-four patients in those selected studies were further investigated, in which 64 of 84 patients (76%) were reported to have SF3B4 pathogenic variant, while the remaining 24% were caused by non-SF3B4 variants (Figure 2A). The current updated analysis found a higher percentage of SF3B4 variant than previously reported.1,4,5 Variants in SF3B4 exon 3 are commonly occurred (20%) (Figure 2A). It should be noted that after the number of variants was adjusted by the number of nucleotides within each exon, the hotspot location was identified in exon 1 (12%, Supplementary Table 2). Among patients with SF3B4 variants, 86% (55 of 64 patients) consisted of NS, while only 14% (9 of 64 patients) were diagnosed with Rodriguez syndrome (Figure 2B). Moreover, we identified that 64% of SF3B4 pathogenic variants belonged to frameshift, followed by nonsense (19%) and variants resulting in loss of initiator methionine (17%) (Figure 2C). We summarized the identified distinct pathogenic SF3B4 variants in Figure 2D, with two common variants, c.1A > G (n = 8 patients) and c.1060dupC (n = 6 patients), located in exons 1 and 5, respectively.

Genetics of Nager syndrome (NS). (A) Variant rate identified in known NS individuals. SF3B4 pathogenic variants contributed to 76% of the cases. Among them, 20% were located in exon 3, followed by 15% in exon 6. (B) Among patients with the SF3B4 variants, 86% were diagnosed with NS and 14% were classified as Rodriguez syndrome. (C) Three common types of variants were identified in patients with SF3B4 variants. Frameshift dominant the cases that contributed 65%, followed by nonsense variants (18%) and loss of initiator methionine (17%). (D) Schematic representation of SF3B4 gene with the variants summarized in this study, which with two common variants, c.1A > G (n = 8 patients) and c.1060dupC (n = 6 patients) were located in exons 1 and 5, respectively.
No Significant Association Between Clinical Features and Variants Within SF3B4 Exons
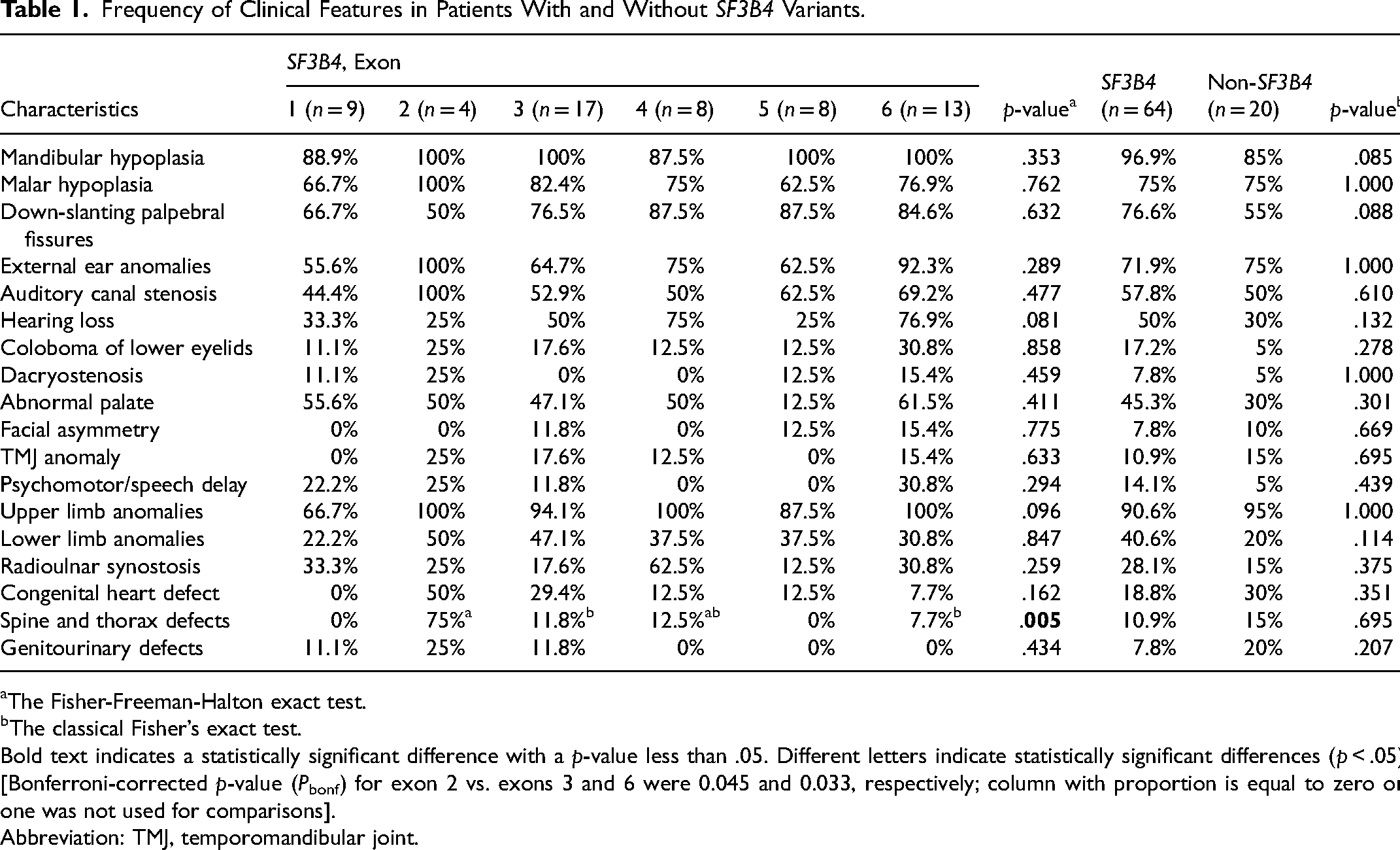
To identify an association between the clinical features and variants within SF3B4 exons, we compared the proportion of one clinical feature in patients with variants in exons 1 to 6. In general, we did not observe any significant association between the clinical features and the six exons studied (p-values for 17 of 18 clinical features were >.05), except patients with variants in exon 2 that exhibited a higher frequency of spinal and thoracic defects (3 of 4 patients, 75%, Table 1). Notably, we identified that congenital heart defect was observed more frequently in patients with variants located in exon 2 (2 of 4 patients, 50%) and 3 (5 of 17 patients, 29.4%).
Frequency of Clinical Features in Patients With and Without SF3B4 Variants.
The Fisher-Freeman-Halton exact test.
The classical Fisher’s exact test.
Bold text indicates a statistically significant difference with a p-value less than .05. Different letters indicate statistically significant differences (p < .05) [Bonferroni-corrected p-value (Pbonf) for exon 2 vs. exons 3 and 6 were 0.045 and 0.033, respectively; column with proportion is equal to zero or one was not used for comparisons].
Abbreviation: TMJ, temporomandibular joint.
No Significant Associations of Clinical Features Between SF3B4 Variants and Non-SF3B4 Variants
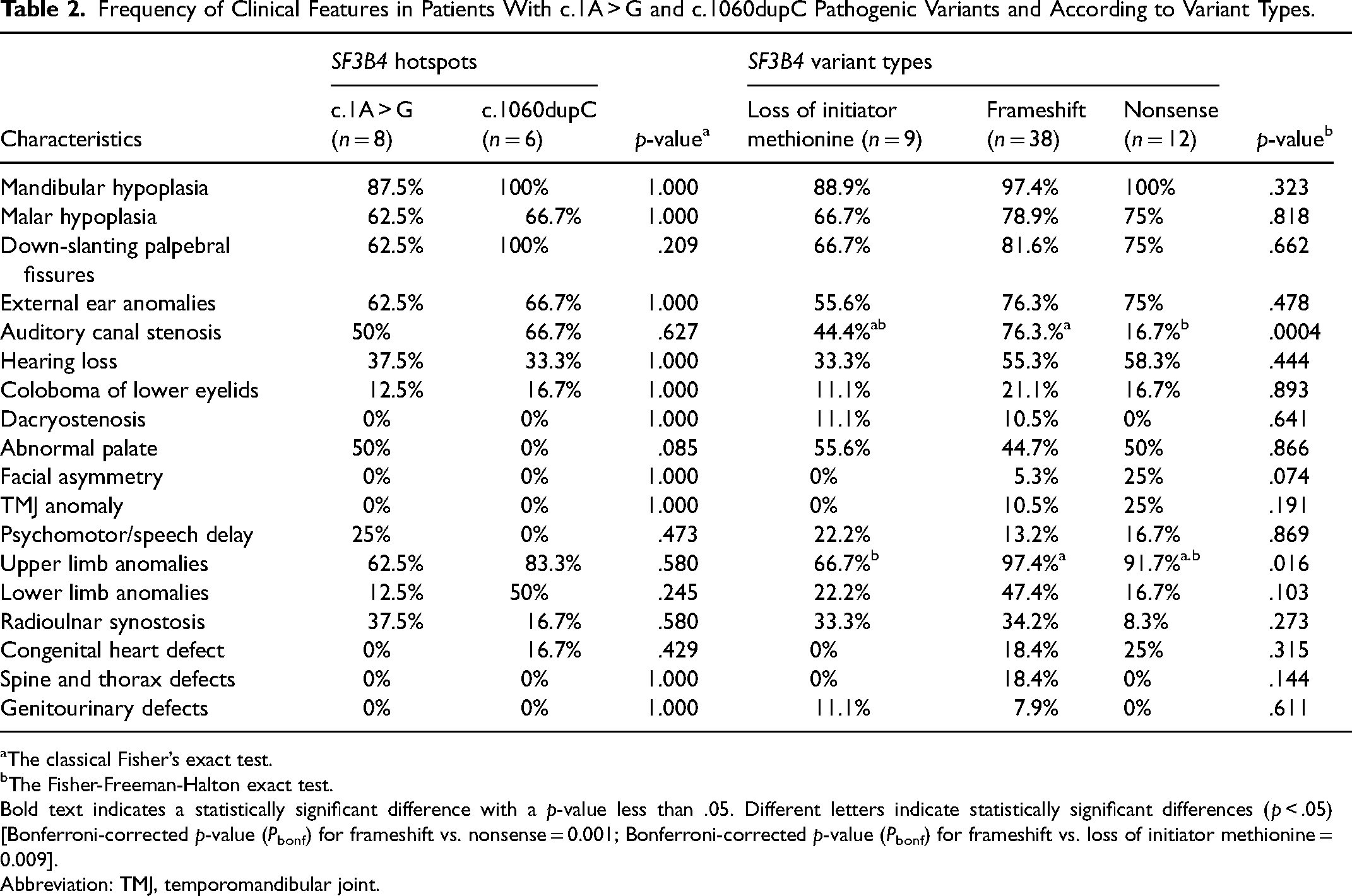
The clinical features in patients with or without SF3B4 variants were compared, and no significant associations (p-values for 18 of 18 clinical features were >.05) were found between the two groups. However, patients with SF3B4 variant tended to develop more severe phenotypes, characterized by a higher occurrence of mandibular hypoplasia and down-slanting palpebral fissures (Table 1). On a closer observation of two SF3B4 variant hotspots, c.1A > G and c.1060dupC, no major phenotypic differences between the two pathogenic variants were found (Table 2). Additionally, the three types of variants in SF3B4 were further compared, and patients with frameshift were likely to have relative severe features of NS such as auditory canal stenosis and upper limb anomalies (Table 2). Lastly, it should be noted that 9 out of 18 lethal cases (50%, Supplementary Table 1) were reported to have frameshift variants.
Frequency of Clinical Features in Patients With c.1A > G and c.1060dupC Pathogenic Variants and According to Variant Types.
The classical Fisher’s exact test.
The Fisher-Freeman-Halton exact test.
Bold text indicates a statistically significant difference with a p-value less than .05. Different letters indicate statistically significant differences (p < .05) [Bonferroni-corrected p-value (Pbonf) for frameshift vs. nonsense = 0.001; Bonferroni-corrected p-value (Pbonf) for frameshift vs. loss of initiator methionine
Abbreviation: TMJ, temporomandibular joint.
SF3B4 Variants with NMD Resulted in Higher Occurrence of Spinal and Thoracic Defects
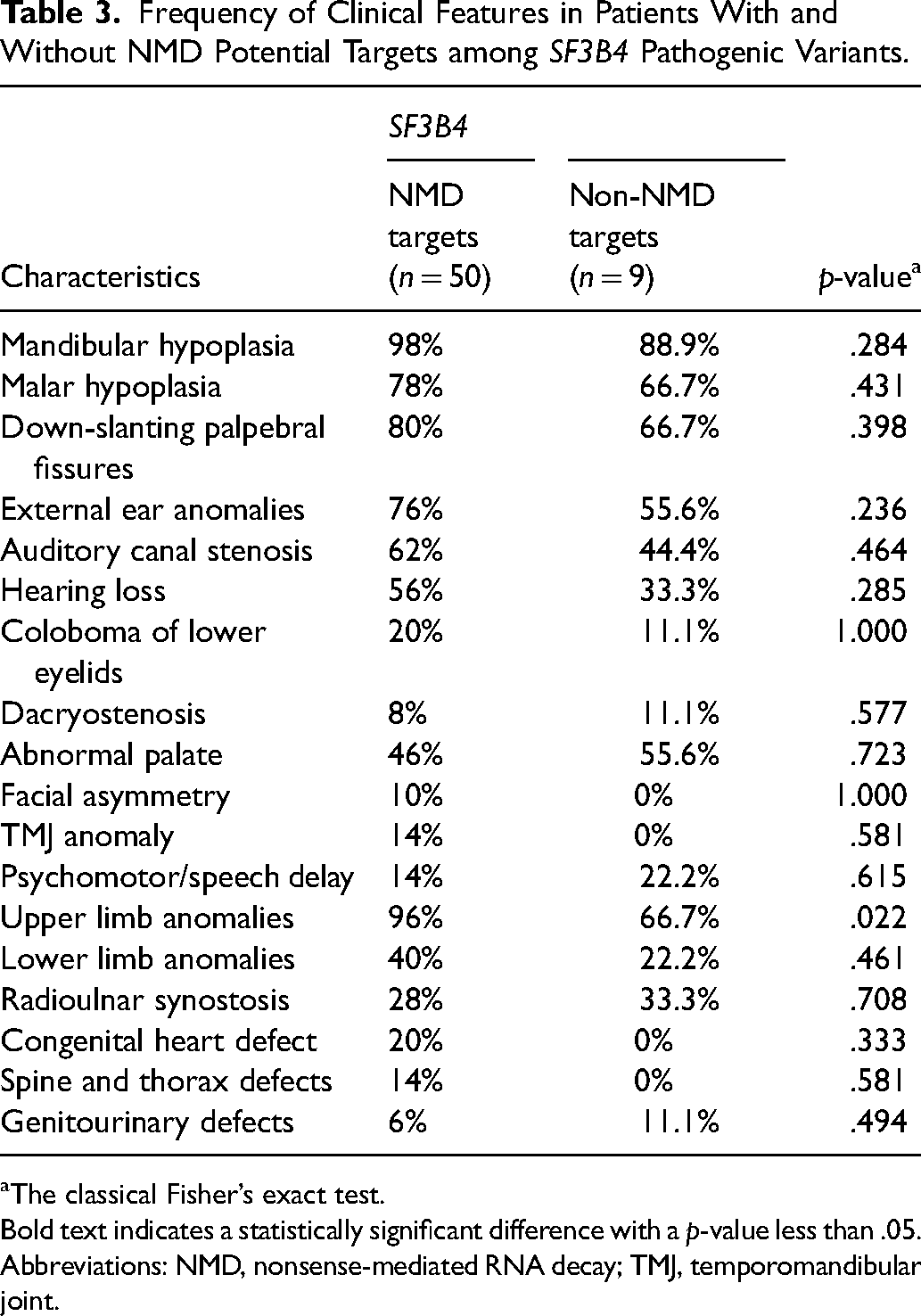
Since most of the SF3B4 variants are predicted to encode a truncated polypeptide chain or an elongated polypeptide with an altered 3′ end in the absence of nonsense-mediated RNA decay (NMD), we then identified the NMD potential targets among SF3B4 variants included in this study. When the proportion of phenotypic features in patients with and without NMD was compared, no significant associations were found between the two groups. However, patients with SF3B4 variants that are predicted to lead to NMD of the transcript could result in more severe phenotypes, such as a higher occurrence of upper limb anomalies (p < .05, Table 3).
Frequency of Clinical Features in Patients With and Without NMD Potential Targets among SF3B4 Pathogenic Variants.
The classical Fisher’s exact test.
Bold text indicates a statistically significant difference with a p-value less than .05.
Abbreviations: NMD, nonsense-mediated RNA decay; TMJ, temporomandibular joint.
Discussion
The pathogenesis of NS is still not fully understood. The formation of proper facial structure requires a highly regulated signaling mechanism, which involves both RNA biogenesis and the neural crest cells. The reported multifunctional roles of SF3B4 provide a hint for dissecting the disease mechanism. The SF3B4 encodes a spliceosome-associated protein, SAP49 that regulates RNA splicing by binding to the pre-mRNA and interacting with other U2 snRNPs. 1 The SAP49 has been shown to inhibit the BMP-mediated osteochondral cell, suggesting its involvement in skeletal development. 5 Furthermore, suppression of neural crest genes (eg sox10, snail2, and twist) that lead to reduce the formation of craniofacial cartilages have been reported in Sf3b4-depleted Xenopus embryos. 25 Further fundamental biological studies could help us to unfold the pathogenesis of the disease.
Haploinsufficiency of SF3B4 is considered to be the major cause of NS, as a result of truncated or altered SF3B4 molecules. 4 Our findings indicated that patients with frameshift variants in SF3B4 tended to have relatively severe NS manifestations, marked by higher proportions of auditory canal stenosis and upper limb anomalies, with 50% of cases being lethal. An animal study shows that the expression of the neural crest genes Sox10 seems to be lower in Sf3b4-depleted Xenopus embryos rescued with mRNA containing human frameshift mutation in exon 2 (c.88delT) than a nonsense mutation in exon3 (c.625C 4 T). 25 Additionally, a study confirmed that depletion of SF3B4 is not only influenced the neural crest formation but also otic development by modulating the expression of several genes responsible for pan-placodal (eg six1, dmrta1, and foxi4.1) and otic vesicle development (eg pax8, tbx2, otx2, bmp4, and wnt3a). 26 Despite the fact that limb anomalies are considered as a typical characteristic of NS, a broader phenotypic spectrum associated with SF3B4 variants has been identified, as such patient harboring substitution variant (c.1168C > T) displays without any limb defects, 24 implying a phenotypic expansion of SF3B4 variant. Therefore, according to the above reasons, it is possible to speculate that craniofacial, otic, as well as extremities development, may be greatly affected in patients with frameshift variants. Indeed, frameshift mutations are considered harmful and among the most deleterious changes to the coding sequence of a protein. 27
In line with previous findings,4,11 our observation strengthens the notion that variants within SF3B4 gene that are predicted to lead to NMD of the transcript could result in more severe acrofacial dysostosis phenotypes. This may be explained by the large degraded mutant transcript of patients with NMD. As a consequence, the small amount of abnormal SAP49 protein produced by the cells may alter the splicing function, although the mechanism remains unclear. Indeed, spliceosome is not only essential in modulating intron splicing but is also a key mediator of alternative splicing. 28 And thus, the clinical variability observed across NS cases could be a result of the aberrant splicing of genes involved in craniofacial and limb development.
On the other hand, there are no distinctive clinical signs between NS individuals with or without SF3B4 variants. Notably, some of the patients without SF3B4 pathogenic variants exhibited variants in EFTUD2,3,4 VWA1, and PYGO2.14,16 Thus, it is possible to hypothesize that other spliceosomal proteins may be involved in the pathogenesis of NS. Further studies with broader genetic testing and larger patient numbers are therefore necessary to fully identify the genetic causes of the disease; while the implementation of model organisms for biological experiments could help us to understand the pathogenesis of NS in the future.
Limitations
Several limitations were identified in this study. First, many of the reported cases did not provide any mutational analysis and complete clinical data. Second, publication bias may be present due to cases with more severe characteristics being commonly published. Thirdly, there is a subjective report regarding clinical manifestations in each of the included studies. Despite said limitations, our study pinpoints that patients with frameshift SF3B4 variants, and predicted to lead to NMD of the transcripts tended to have more severe clinical manifestations observed in NS.
Supplemental Material
sj-xlsx-1-cpc-10.1177_10556656221089156 - Supplemental material for SF3B4 Frameshift Variants Represented a More Severe Clinical Manifestation in Nager Syndrome
Supplemental material, sj-xlsx-1-cpc-10.1177_10556656221089156 for SF3B4 Frameshift Variants Represented a More Severe Clinical Manifestation in Nager Syndrome by Zulvikar Syambani Ulhaq, Gita Vita Soraya, Lola Ayu Istifiani, Syafrizal Aji Pamungkas and William Ka Fai Tse in The Cleft Palate Craniofacial Journal
Supplemental Material
sj-docx-2-cpc-10.1177_10556656221089156 - Supplemental material for SF3B4 Frameshift Variants Represented a More Severe Clinical Manifestation in Nager Syndrome
Supplemental material, sj-docx-2-cpc-10.1177_10556656221089156 for SF3B4 Frameshift Variants Represented a More Severe Clinical Manifestation in Nager Syndrome by Zulvikar Syambani Ulhaq, Gita Vita Soraya, Lola Ayu Istifiani, Syafrizal Aji Pamungkas and William Ka Fai Tse in The Cleft Palate Craniofacial Journal
Footnotes
Author’s Contribution
Conceived the study: Z.S.U., W.K.F.T.; Collected the data: Z.S.U., L.A.I., S.A.P.; Performed the analysis: Z.S.U., G.V.S., L.A.I., S.A.P.; Wrote the paper: Z.S.U., G.V.S., W.K.F.T.; Supervision: W.K.F.T.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval and permission are not required because this study includes no confidential personal data and patient interventions. This study is a meta-analysis based on published eligible studies of genotypic and phenotypic findings in Nager syndrome patients.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.