
Abstract
Objective
Paired box 7 (PAX7) has been considered as a candidate gene for non-syndromic cleft lip with or without palate (NSCL/P). However, there is no research for the XXX, and previous studies concentrated on limited variants. This study aimed to conduct sufficiently dense and powerful scans of variants at PAX7 and explored the roles of variants at PAX7 in NSCL/P among the XXX.
Design
Targeted region sequencing was performed to thoroughly screen variations, followed by a two-phase association analysis. 159 NSCL/P cases and 542 controls were analyzed in phase 1. Then in phase 2, the validation study was performed using 1626 cases and 2255 controls. We also explored the roles of variants at PAX7 gene in NSCL/P subtypes. Additionally, indirect associations were found by calculating LD and haplotypes.
Setting
The study was conducted in XXX.
Patients, participants
159 NSCL/P cases and 542 controls were analyzed in phase 1. Then in phase 2, the validation study was performed using 1626 cases and 2255 controls.
Interventions
Blood samples were collected.
Main outcome measures
To explore the association analysis between variants at PAX7 and NSCL/P in XXX.
Results
The results showed that rs2236810, rs114882979 and rs2236804 were significantly associated with NSCL/P, which were predicted to have regulatory functions. Besides, variants at PAX7 function differently in the NSCL/P subtypes. We also discovered a PAX7 missense variant, NM_001135254 p.A369 V (NM_002584.2:c.1106C > T).
Conclusions
In summary, we confirmed 3 SNPs at PAX7 were significantly associated with NSCL/P in XXX and identified a missense variant, NM_001135254 p.A369 V (NM_002584.2:c.1106C > T).
Introduction
The most common birth abnormality affecting the craniofacial region in humans is non-syndromic cleft lip with or without palate (NSCL/P). Based on the differences in affected anatomical structures, NSCL/P can be divided into two types: non-syndromic cleft lip only (NSCLO) and non-syndromic cleft lip with cleft palate (NSCLP). 1 In China, according to a recent study, the pooled prevalence rate of NSCL/P was 1.67‰, varying among provinces, which was rather high among worldwide. In different subtypes, the pooled prevalence estimate was 0.56‰ for NSCLO and 0.82‰ for NSCLP. 2 Patients with NSCL/P experience issues with speech, hearing, appearance, psychology. This requires multidisciplinary care from birth to adulthood, long-term negative effects on the patient's family and society. 3 Therefore, it is necessary to screen out more effective therapeutic targets and fully elucidate the pathogenesis of this congenital disorder for better prevention and treatment.
NSCL/P is a disease of multifactorial etiology involving genetic and environmental factors. With the advent of genome-wide association studies (GWASs) and genetic linkage studies, several studies have identified multiple loci influencing the risk of NSCL/P,4–8 including PAX7.4,6,9 Subsequent studies with different genetic backgrounds indicated population-specific relationships and subtype-specific relationships between PAX7 and NSCL/P. A study, performed by Sull et al., exploring PAX7 variants in four populations excluding the Chinese mainland showed a strong parent-of-origin effect, while a Baltimore analysis produced contradicting findings.7,10 Furthermore, studies have shown that PAX7 behaved differently in NSCL/P subtypes. PAX7 was nominally associated with NSCL/P but not with NSCPO in Sub-Saharan African Population, 11 whereas PAX7 was found to be connected with both NSCLO and NSCLP in European populations. 12
However, no study has looked into whether variations at PAX7 affect disease risk in the XXX population, and previous studies only focused on the sites involved in GWAS. The genetic association between PAX7 and NSCL/P in the XXX population remains unknown, and the genetic associations between PAX7 and NSCL/P have not been thoroughly investigated.
The present study aimed to explore PAX7's potential contribution to the etiology of NSCL/P and its subtypes in the XXX population. We intend to undertake a deep screening targeting PAX7 to fully dig into variants at PAX7 and uncover susceptibility SNPs or indels through a relatively large-scale trial of genetic association analyses.
Materials and Methods
Study Population
Initially, the study recruited 159 NSCL/P patients from XXX from 2011 to 2016 (containing 79 cases of NSCLO and 80 cases of NSCLP). Whole genome sequencing data of 542 normal controls used in the first phase association analysis were obtained from Novogene internal database (http://www.novogene.com/). In the second phase, we extracted genotype data from samples collected during our prior study's GWASs.6,13 Before being enrolled in the study, all study participants—or their legal guardians—gave written informed consent. Clinicians with experience in surgery, pediatrics, and genetics made clinical diagnosis.
Inclusion criteria:1.Patients and their parents are XXX population; 2.Patients with cleft lip and palate phenotype and no other systemic diseases; 3.No other hereditary diseases in the patient's family.
Exclusion criteria:Contrary to any one of the inclusion criteria.
Extraction of Genomic DNA
Genomic DNA of 159 cases was extracted from venous blood by protein precipitation method and dissolved in Tris-ethylene diamine tetraacetic acid buffer as previously reported. 14 The purity and concentration of DNA were detected by NanoPho-tometer® spectrophotometer (IMPLEN, CA, USA) (OD260/OD280). Then, acceptable DNA samples were kept at −80 °C with an OD value of roughly 1.8.
Targeted Sequencing at PAX7 Gene and Genotyping
Firstly, we performed targeted sequencing in 159 patients with NSCL/P. Based on the LD structure in Han Chinese in Beijing, China (CHB)/Japanese in Tokyo, Japan (JPT) HapMap, we chose haplotype around PAX7 spanning chr1:18957340-19075360 and used Agilent liquid capture system (Agilent Sure Select XT Custom Kit) to enrich and capture the targeted sequence from 1.0 μg genomic DNA according to the manufacturer's protocol. PCR products were sequenced by ABI PRISM 3730 DNA Sequencer (Functional Biosciences, Inc., Madison, WI) to identify mutations, the data were analyzed by Sequence Scanner v1.0. For quality control, 15 we obtained high-quality variants which satisfied: 1) a call rate of >95%; 2) Hardy Weinberg test p-value of >0.01. Sanger sequencing was performed for variants validation and segregation analysis.
Data Processing and Quality Control
All subsequent bioinformatics analyses were based on clean data with high quality. To obtain the original mapping result in binary alignment map (BAM) format, valid sequencing data were mapped to the reference genome (GRCh37/hg19) by Burrows-Wheeler Aligner (BWA)software. Subsequently, Samtools and Picard were utilized to sort bam files, mark duplicate reads, and generate a final bam file. Samtools mpileup and bcf tools were used to call variants and identify SNPs, indels. Annotation for VCF (Variation Call Format) file was generated through ANNOVAR. Various databases, such as dbSNP, 1000 Genome, ExAC, CADD, and Human Gene Mutation Database (HGMD), were retrieved to obtain the variation position, variation type, conservative prediction, and other information. Gene transcript annotation databases, such as Consensus coding sequence (CDS), RefSeq, Ensembl, and UCSC, are also applied to annotate exonic variations to determine amino acid alternation. Finally, functional prediction for the nonsynonymous single nucleotide variants (SNVs) was performed by PolyPhen2, SIFT, MutationTaster, CADD.A series of quality control checks and filtering strategies were performed. The quality control for the raw sequencing data included filtering adapter-related reads, reads containing N, and low-quality reads. The quality control of the genotyping included filtering of genotyping rate less than 95%, tri-allelic or tetra-allelic (di-allelic variants only), MAF (minor allele frequency) above 0.01, and HWE (Hardy–Weinberg equilibrium) P-value more than .01.
Statistical Analysis
To compare the allele frequency of the SNPs between the patients and healthy controls in the first phase, the Chi-square test was used after excluding SNPs that departed from Hardy-Weinberg equilibrium (HWE) and/or had MAF (minor allele frequency) 1%. A P-value of ≤.05 was taken as significant. 16 Unconditional logistic regression analysis was used to evaluate the association between allele frequencies and risk of NSCL/P in the second phase and subtype association analysis. After using the Bonferroni multiple corrections, the significant threshold was taken as 0.00079 (P = .05/63). Additionally, 95% confidence interval (95% CI) and odds ratios (OR) were calculated. Data analysis was done using PLINK. Sliding window-based haplotype association tested with Generalized Linear Models (GLMs) were performed using a logistic regression model to examine the effect of a haplotype of various lengths on the risk of NSCL/P and its subtypes. 17 Interactions between SNPs were analyzed to explore whether the SNPs functioned independently using the MB-MDR package for R.
Functional Prediction
To investigate the function of SNPs, we performed bioinformatics analysis using three online prediction websites: HaploReg, RegulomeDB and ChromHMM. We explored the chromatin state in which the SNPs are located by ChromHMM. ChromHMM is an integrated database containing multiple chromatin datasets such as ChIP-seq data for universal annotation of the human genome.18,19 As the chromatin state annotation results suggested that these SNPs may be located within regulatory elements, we further explored whether they affect transcription factor binding affinities using HaploReg and RegulomeDB. HaploReg was employed to explore noncoding genomic annotations for variants and to identify their potential causal relationship with the pathogenesis of the disease. 20 RegulomeDB is a database that provides functional annotations of SNPs, including data on chromatin structure, methylation, protein motifs, and binding. 21
Results
Characteristics of Study Participants
In this study, we enrolled 159 patients for targeted sequencing and first-phase genetic association analysis (Table 1). Of the 159 patients with NSCL/P, 79 had NSCLO, and 80 had NSCLP. There was no significant difference in sex between the cases and the controls. In addition, 1626 patients with NSCL/P and 2255 controls were enrolled in the second phase (Table 1). Also, no significant differences were found between the patients with NSCL/P and control groups with respect to age or sex.
Characteristics of NSCL/P Patients and Control Group.

Note: NSCLO, non-syndromic cleft lip only; NSCLP, non-syndromic cleft lip with cleft palate; NSCL/P, non-syndromic cleft lip with or without palate.
Discovery and Validation of NSCL/P-Associated Common Variants
Comparing the frequency of each variant in cases (n = 159) versus controls (n = 542), we identified 63 variants that were significantly associated with susceptibility to NSCL/P (P < .05, Supplementary Table 1). There were 37 SNPs found in intronic regions, 24 SNPs found in intergenic regions, one SNP found in the 5'UTR, and one SNP in the upstream. Variant rs609959 in intron 7 showed the strongest association (P = 1.54E-04; Figure. 1).
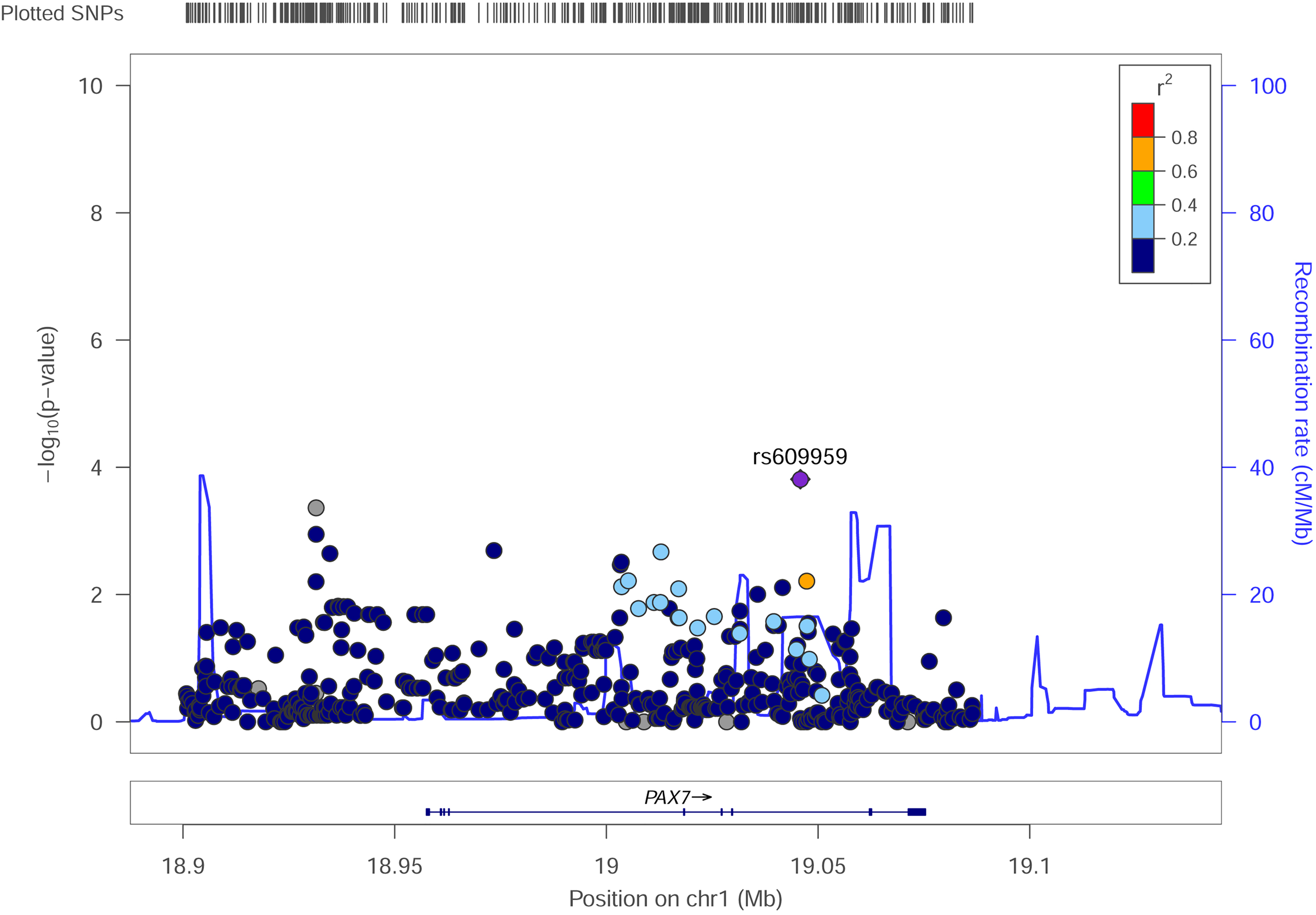
Regional association plot of SNPs at PAX7. SNP related to NSCL/P is colored purple, and different colors represent different LD with purple SNPs.
In the second phase, the results revealed that, of the 63 SNPs, 3 SNPs were significantly associated with NSCL/P (Figure 2). The most significant SNP is rs2236810 with a P-value of 1.97 × 10−5. Analysis of SNP-SNP interactions indicated that these SNPs contribute independently to NSC/LP (Supplementary Table 2).
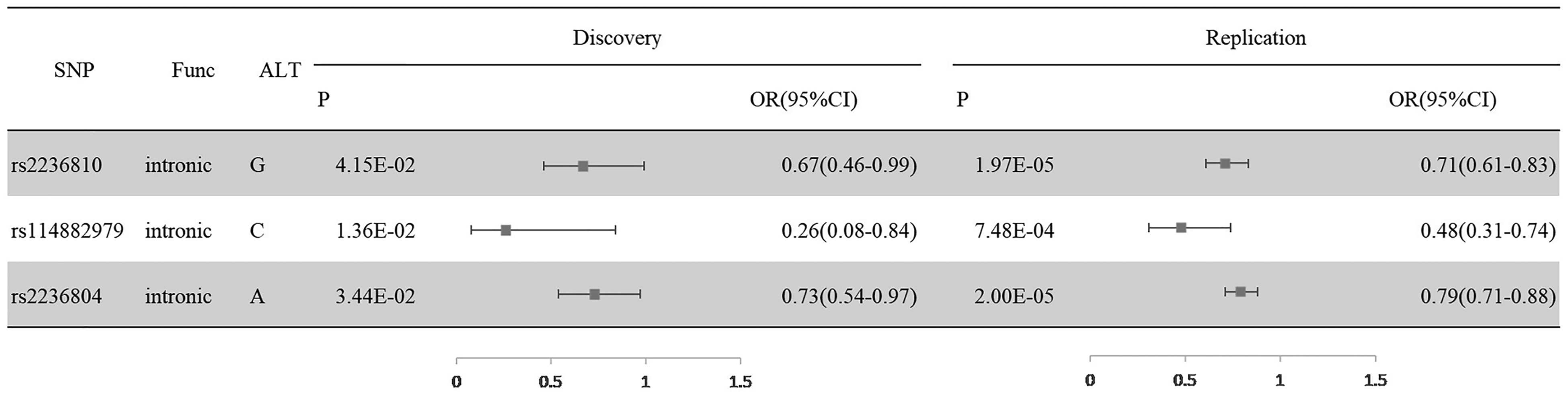
Replication of the NSCL/P-associated SNPs. Note: SNP, single nucleotide polymorphism; NSCL/P, non-syndromic cleft lip with or without palate; NSCLP, non-syndromic cleft lip with palate; NSCLO, non-syndromic cleft lip only; Alt, alternate allele; P, P value; OR, odds ratio; 95%CI, 95% confidence interval.
In Silico Bioinformatics Analysis
Since the 3 NSCL/P-associated SNPs were all mapped to the intronic region, we speculated that their influence on NSCL/P pathogenesis may be mediated through transcriptional regulation. Therefore, we used the ChromHMM to detect whether SNPs were located in regulation elements. According to ChromHMM, all the 3 SNPs serve as transcriptional regulators in different tissues, such as enhancers, implying that they may act as functional variants in the regulatory network and ultimately result in NSCL/P (Supplementary Table 3). Motif analysis by RegulomeDB database revealed that 3 SNPs bind to transcription factors (Figure 3). We also retrieved HaploReg to detect whether SNPs affect the binding affinities of transcription factors. 2 SNPs showed allele-specific transcription factor binding affinities (Table 2). Notably, some of these affected transcriptional factors have been previously reported to be associated with NSCL/P.
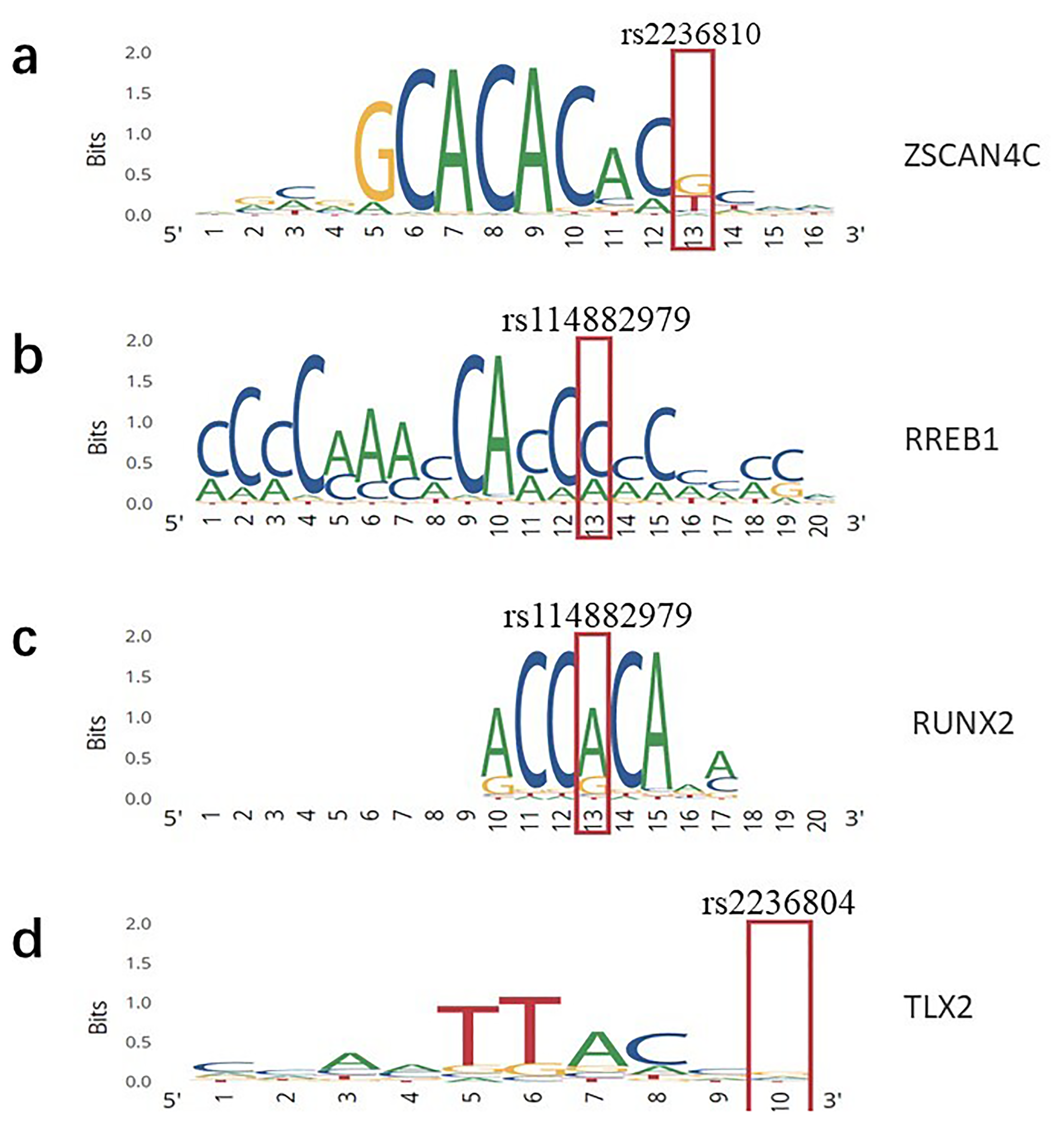
Position weight matrix (PWM) model for representing transcription factor (TFBS) binding site motifs. The sequence logo shows the changes in predicted transcription factor binding sites.
Allele-Specific Transcription Factor Binding Affinities.
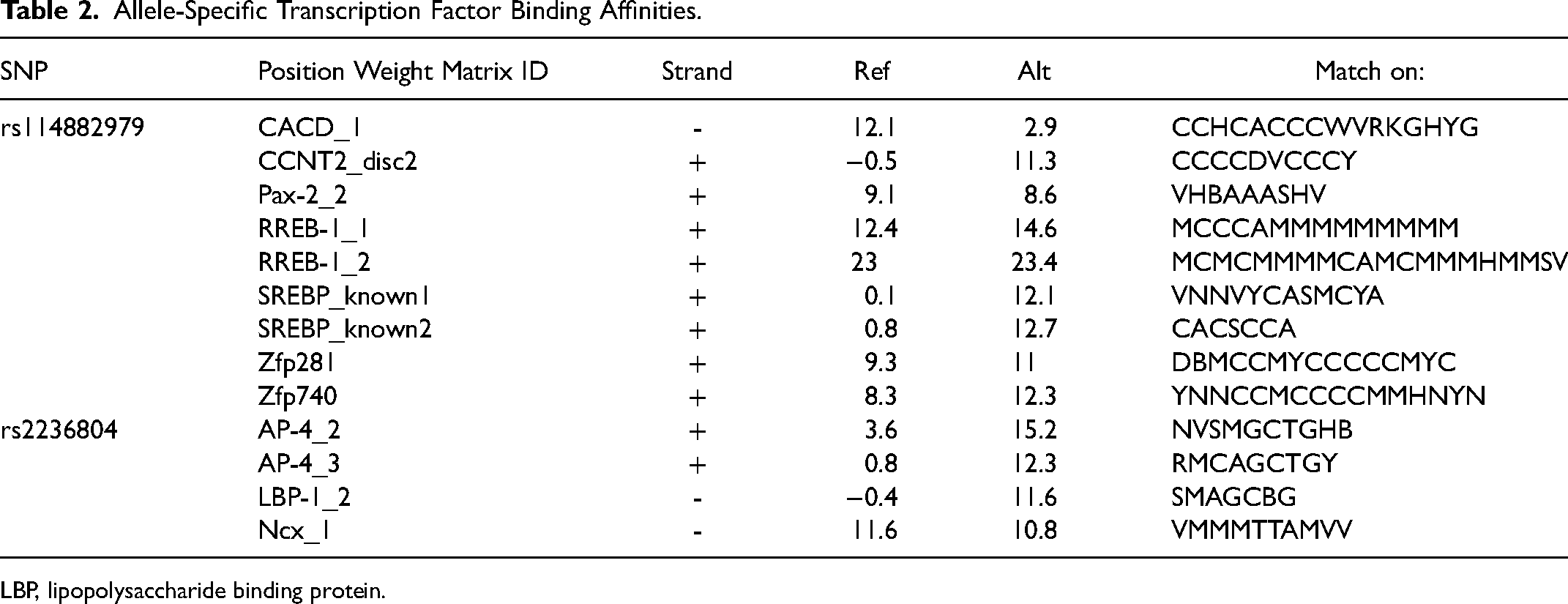
LBP, lipopolysaccharide binding protein.
Linkage Disequilibrium (LD) and Haplotype Analysis of NSCL/P-Associated SNPs at PAX7
In order to test whether the selected SNPs located on the same chromosome were also in the same LD block, we performed the pairwise LD analysis using the Haploview program. 22 The results showed that the pair of rs2236810, rs114882979 and rs2236804 were all linked (D’ > 0.94, r2 = 0.33, Supplementary Figure 2a). According to the sliding window haplotype analysis (Supplementary Figure 2b, Supplementary Table 5), haplotypes at PAX7 were associated with the risk of NSCL/P. Individual carriers of haplotype AAG (rs2236810, rs114882979 and rs2236804) at the PAX7 gene may have a significantly lower risk of NSCL/P (P = 3.67 × 10−7).
Heterogeneity of the NSCL/P Subtypes
Due to the similar embryonic development and epidemiological characteristics, NSCLO and NSCLP are collectively referred to as cleft lip with or without cleft palate (NSCL/P). However, in recent years, it has been found that each subtype actually has a different genetic background.23,24 We conducted a subtype association analysis to confirm if these identified 63 SNPs are risk factors in different subtypes. Interestingly, the SNPs associated with NSCLP and NSCLO did not overlap (Table 3). Rs582451, rs2236810, rs2236804 were specifically associated with NSCLP, while rs114882979 was specifically associated with NSCLO. Association between rs582451 and NSCL/P was observed in the first phase, but it failed to pass the second phase of validation. The P-value for the association of rs582451 with NSCL/P in the second phase is 8.03 × 10−2.
Subtype-Specific Associations.
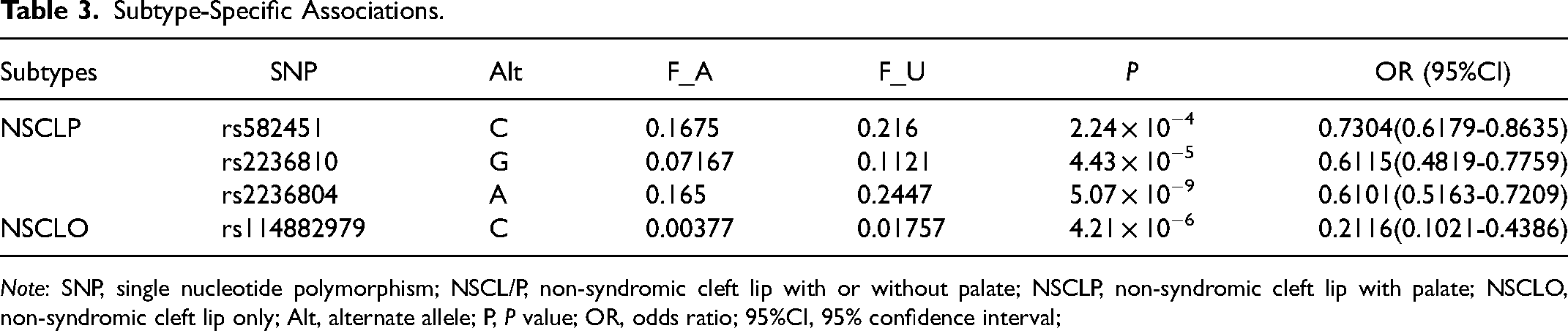
Note: SNP, single nucleotide polymorphism; NSCL/P, non-syndromic cleft lip with or without palate; NSCLP, non-syndromic cleft lip with palate; NSCLO, non-syndromic cleft lip only; Alt, alternate allele; P, P value; OR, odds ratio; 95%CI, 95% confidence interval;
Rare Variant
Besides, we discovered a PAX7 missense variant, NM_001135254 p.A369 V (NM_002584.2:c.1106C > T), during the sequencing process. The patient and his parents underwent Sanger sequencing targeting the variant for validation. The case inherited this missense variant from his unaffected mother (Supplementary Figure 1).
Discussion
The paired-box (PAX) gene family played an essential role during early embryonic development, particularly during neural crest development, which was closely associated with cranial and maxillofacial development. 25 As a member of the PAX family, PAX7 has also been widely reported on the relationship between neural crest development and NSCL/P. Basch et al. revealed PAX7 as an early marker required for avian neural crest formation. 26 Pax7 mutant mice showed facial skeletal structures malformation that might be related to neural crest cell defect during embryonic development. 27 Pax3/Pax7 double mutant mice showed impaired function against environment-related teratogenesis, and exhibited more severe facial morphogenesis defects than either mouse with impaired Pax3 or Pax7 functions. 28 In vivo transgenic manipulation revealed that Pax3 and Pax7 regulated stem cell behavior and affected tissue differentiation by interacting with fibroblast growth factors pathway, which was a key pathway in proliferation and differentiation during embryogenesis. 29
Our understanding of the mechanisms behind NSCL/P has significantly increased as a result of recent developments in laboratory mice, but genetic investigation of PAX7 was insufficient for the restricted sites and populations. Determining the association between genetic variation and phenotype is a pivotal step in studying the mechanism of NSCL/P, laying the foundation for studying drug therapies and biomarkers. Adequate genetic analysis of genes is necessary for detecting and preventing NSCL/P. Thus, we carried out a relatively large-scale trial of genetic association analyses in the XXX population to gain a more sophisticated understanding of the genotypic spectrum of NSCL/P.
In this study, 159 NSCL/P patients underwent targeted sequencing, a method that thoroughly screening targeted regions and even reliably detects low-frequency variants there.30–32 Then, 63 SNPs were identified to be significantly associated with NSCL/P in the XXX population, which may contain false signals due to mixed samples of different sub-types. Therefore, subsequent validation using a larger sample size was conducted, and 3 of the 63 SNPs also showed significant association with NSCL/P. Functional predictions were made using different online databases. We also conducted an independent genetic association study to learn more about the function of these SNPs at PAX7 in NSCLP and NSCLO, and the outcomes showed subtype heterogeneity. Based on the analysis of SNP, haplotype analysis was carried out. Haplotypes in PAX7 modify the risk of NSCL/P in the XXX population. Our study thoroughly explored the relationship between PAX7 and NSCL/P in the XXX population, and expanded the understanding of disease etiology.
A strategy to advance biological insight is to annotate variants and map them to genes using functional genomic resources. 33 The SNPs we identified all located in the intronic regions of PAX7, and introns may increase gene expression by affecting the stability of transcription, the efficiency of mRNA translation, and nuclear export.34,35 Thus, we performed functional predictions and found that these SNPs were associated with genes contributing to the development of cleft lip and palate. For example, motif analysis revealed that rs114882979 might bind to RUNX family transcription factor 2(RUNX2) and Ras-responsive element-binding protein 1(RREB1). Loss of Runx2 in cranial neural crest (CNC)-derivatives results in soft palate muscle defects,36,37 and aberrant RREB1-mediated Ras signaling might play a crucial role in the pathogenesis of cleft palate. 38 Overall, functional predictions imply that SNPs may have an impact on disease progression via altering transcriptional regulation.
A population-based study by Uribe et al. revealed that SNPs within PAX7 had significant associations with both NSCLO and NSCLP subgroups. 12 Inconsistent with Uribe et al., our study showed that these loci play different roles in various sub-types. Results revealed that rs582451, rs2236810 and rs2236804 were significantly associated with NSCLP, not with NSCLO. Rs114882979 were significantly associated only with NSCLO. Comparing the SNPs studied in the present study with other studies worldwide, one of the most interesting SNP was rs742071, an intronic varian in which association with NSCL/P was confirmed in different studies.4,7,9,11,12,39 Rs742071 was suggested to be a tag SNP representing a still unknown functional PAX7 variant associated with the risk of NSCL/P, while it was not identified in our study. Why these SNPs were found associated with the present population, while not with others? We took into account possible factors such as genetic heterogeneity, complexity of the disease, geographical distribution, lifestyle, economic differences, differences in research methods, and collection of experimental samples. This study confirmed the distinct roles of PAX7 in NSCLO and NSCLP, but the different internal connections among them need to be further explored.
In conclusion, we revealed the association between polymorphism of PAX7 gene and NSCL/P in the XXX population and identified PAX7 missense variant, NM_001135254 p.A369 V (NM_002584.2:c.1106C > T). Our research contributed to a better understanding of the role of PAX7 in the occurrence of NSCL/P. However, this study still has some limitations. The results of this study were not validated in cellular or animal models and it needs to be validated in other large sample population studies. Further functional analysis is needed to explore the mechanism by which nucleotide variants in the PAX7 gene may increase susceptibility to NSCL/P. And how PAX7 gene is involved in the development of NSCL/P still requires functional studies to reveal its role in the regulatory network.
Conclusion
In summary, we confirmed 3 SNPs at PAX7 were significantly associated with NSCL/P in the XXX population and identified a PAX7 missense NM_001135254 p.A369 V (NM_002584.2:c.1106C > T).
Supplemental Material
sj-docx-1-cpc-10.1177_10556656231163398 - Supplemental material for Integrated Analysis of the Association Between Variants at PAX7 and NSCL/P in the Han Population
Supplemental material, sj-docx-1-cpc-10.1177_10556656231163398 for Integrated Analysis of the Association Between Variants at PAX7 and NSCL/P in the Han Population by Cheng-Wei Yang, Yue You, Jia-lin Sun, Bing Shi and Zhong-Lin Jia in The Cleft Palate Craniofacial Journal
Footnotes
Acknowledgements
The authors thank all the participants who donated samples in this study. All authors gave their final approval and agree to be accountable for all aspects of the work. This project was supported by the National Science Funds of China (No. 81600849) and the National Precision Medicine Project (No.2016YFC0905203).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the The National Science Funds of China, (grant number 81600849).
Ethical Approval
All the participants or their guardians need to write informed consent before enrolled into the study and the study protocol was approved by Hospital Ethics Committee of West China Hospital of Stomatology, Sichuan University (WCHSIRB-D-2016-012R1).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.