
Abstract
Objective
To compare the rates of selected nervous system, cardiovascular, and otologic abnormalities in patients with and without Treacher Collins Syndrome (TCS).
Design
Retrospective TriNetX platform cohort study.
Setting
Aggregated and deidentified electronic health record (EHR) data from across the United States.
Patients, Participants
Patients with TCS (n = 1,114) and a propensity matched control cohort without TCS (n = 1,114 matched from n = 110,368,585).
Main Outcome Measured
Prevalence and relative risk (RR) of selected diagnoses in a propensity-matched cohort.
Results
The RR of congenital malformations of the circulatory system in patients with TCS was 8.5 (95% CI 4.44-16.28). Patients with TCS also had higher rates of otologic abnormalities including conductive hearing loss (RR 44, 95% CI 24-83) and nervous system disorders including movement disorders (RR 2.60, 95% CI 1.27-5.50) and recurrent seizures (RR 4.2, 95% CI 2.12-8.33).
Conclusions
We found a significantly elevated risk in TCS patients within all three systems. We postulate that the nervous system effects may be the result of one of the TCS-linked genes, for which a mutation has also been associated with progressive ataxia, cerebellar atrophy, hypomyelination, and seizures. As the previously-identified causal genes influence neural crest cells that form the head and face, these cells may also populate cardiac structures, resulting in cardiovascular abnormalities. Finally, the characteristic craniofacial abnormalities identified in TCS impair hearing and are associated with increased risk of otitis media. Our findings may help researchers to hypothesize the function of the genes underlying TCS, as well as to inform the care of affected individuals.
Introduction
Treacher Collins syndrome (TCS), a kind of mandibulofacial dysostosis, is a rare genetic condition characterized by abnormalities of the head and face including underdeveloped zygomatic bones and mandible, downward slanting palpebral fissures, and external ear abnormalities. Individuals may experience hearing loss, and zygomatic and mandibular hypoplasia can result in feeding and respiratory difficulties. However, phenotypic expression varies significantly: cleft palate, choanal stenosis, and vision loss are also reported, though less commonly.1
Although likely first described in 1846 by others, the ophthalmologist Dr. Edward Treacher Collins is credited for the discovery of the condition in 1900. Its prevalence is estimated at approximately 1 in 50,000 individuals; however, due to the great variation in phenotype and severity, some individuals certainly go undiagnosed. Cases exist of mildly affected previously undiagnosed individuals eventually genotyped due to the birth of a more severely affected relative.2
Several genetic variants are known to cause the condition, including the autosomal dominant mutations of TCOF1, POLR1D, or POLR1B, or, less commonly, the autosomal recessive mutation of POLR1C.3 Only around 40% of cases are thought to be inherited; the other 60% arise from de novo mutations.4 These genes are expressed in neural crest cells and their products are involved in ribogenesis,3 and, given their involvement in TCS, are thought to play a role in the development of the cartilage, bone, and tissue of the head and face.4
The TCS phenotype is traditionally described as being limited superficially to the head and face. As our understanding of both the genetic and phenotypic variation increases, new findings arise, such as delayed motor development which affected 12% of individuals in a German cohort of 46 patients.2 These recent discoveries indicate there is more to be learned about TCS and possibly associated extracraniofacial anomalies. Our ability to quantify other anomalies, however, is limited by the rarity of the diagnosis, resulting in small sample sizes in existing studies.
The present study aims to investigate the epidemiology of cardiovascular and nervous system disorders among individuals with TCS. Due to its characteristic craniofacial effects, we will also provide an estimate of the more-commonly seen otologic complications. Findings will inform care for children and adults with diagnosed or suspected TCS, emphasizing the importance of monitoring for such conditions in this population. Furthermore, as we still seek to fully understand the genetic underpinnings of TCS, these findings may help researchers to hypothesize the function of the genes known to be mutated in this condition.
Methods
A retrospective cohort design was implemented for this study using the “Research” network of the TriNetX platform, a federated health database that aggregates electronic health record (EHR) data across health care organizations (HCOs) globally. The TriNetX research network combines EHR data from over 70 organizations and over 100 million patients. TriNetX has been used in a number of other studies to look at population level statistics.5,6 TriNetX only contains de-identified data and has been deemed exempt from the Western Institutional Review Board (IRB) by a qualified expert as defined in Section §164.514(b)(1) of the HIPAA Privacy Rule. Patient consent was not applicable and not needed. Searches were performed on May 1, 2023.
Patients of all ages were included. Patients in the TCS group were defined as those with at least one encounter diagnosis of TCS, identified via the ICD 10 code Q75.4 for mandibulofacial dysostosis. Although this ICD-10 code encompasses all forms of mandibulofacial dysostosis, Treacher Collins Syndrome is the most common,7 and there is not a more specific code for TCS. Patients in the control group were defined as those without any encounter diagnosis of TCS. Although TriNetX is unable to propensity match with a cohort as large as our control group, we attempted to randomly reduce our control cohort via the additional requirement of an ambulatory visit within three months of our search. This reduced our control cohort to 5,828,462 patients. Using this new group, we propensity matched for age, race, and ethnicity. Our final cohorts were:
TCS group, n = 1,114 Initial control cohort of patients without TCS, n = 110,368,585 Patients without TCS propensity matched for age, race, and ethnicity (control), n = 1,114
Relative risks were calculated using a 95% confidence interval (CI). Demographic data was analyzed via two sample Z-tests for continuous variables and chi-square tests for categorical variables, where significance was defined as p < 0.05.
In TriNetX, diagnoses affecting at least 1 but fewer than 10 patients in a cohort is automatically rounded to 10 to preserve anonymity. Within our tables, diagnoses with at least 1 but fewer than 11 patients are depicted with an asterisk.
Results
Demographic Data
Prior to reduction and propensity matching, TCS patients were significantly younger (p < 0.00001) and significantly more patients with TCS were of white race (p < 0.0001); however, a significantly larger percentage of the control cohort were of unknown race (39% vs 21%, p < 0.0001), which may falsely skew demographic data. There were no significant differences in sex between groups with and without TCS (53% vs 54% female, p = 0.51) (Table 1).
Demographic characteristics for patients with and without Treacher Collins Syndrome (TCS) prior to and after propensity matching for age, race, and ethnicity.
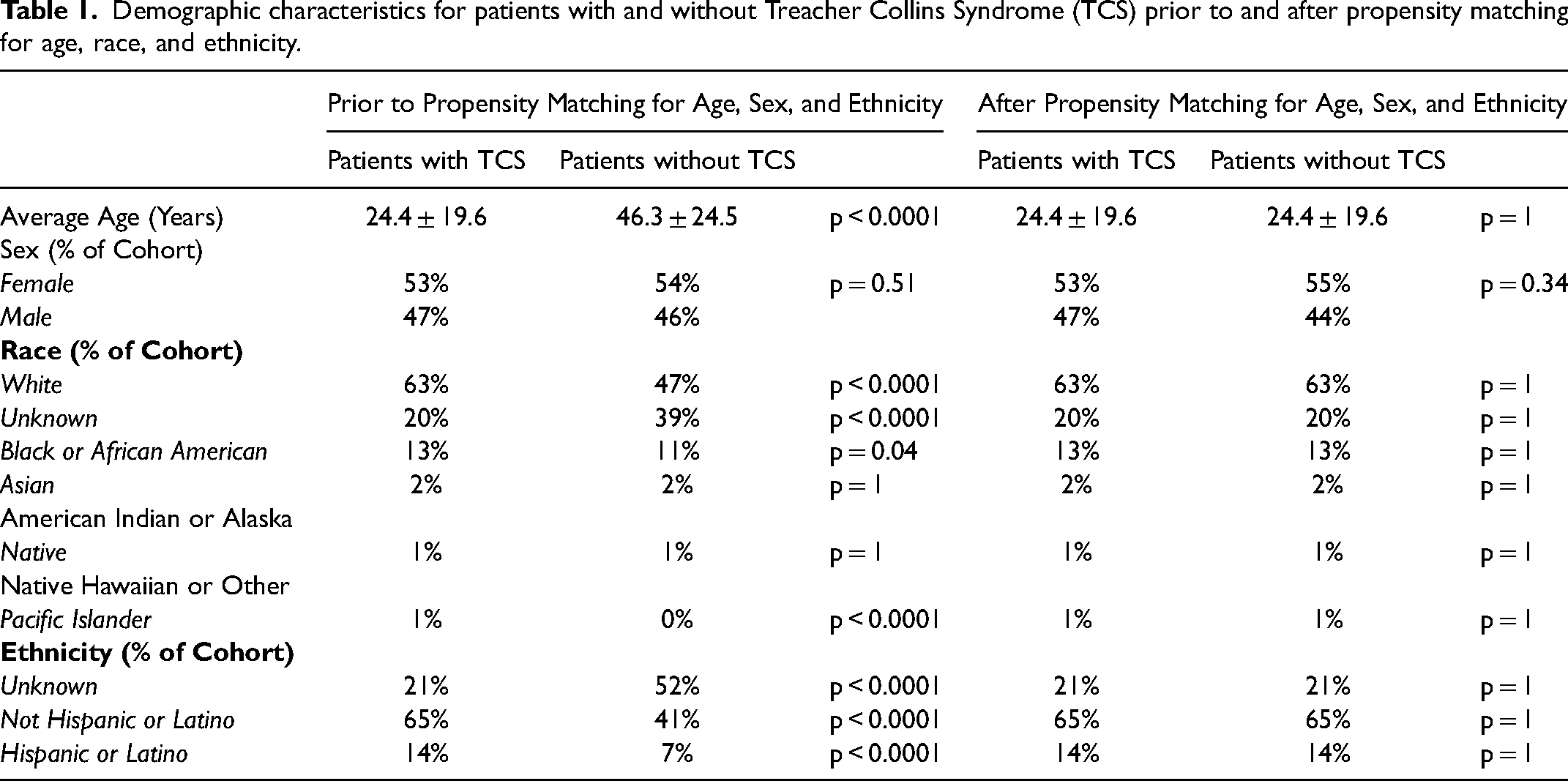
After reduction and propensity matching, there were no differences in race, ethnicity, or average age between our cohorts (p = 1 for all). There was no significant difference in sex between groups (p = 0.34). The new mean ages were 24 ± 19.6 years.
Systems Data after Propensity Matching
After propensity matching, patients with TCS were found to have an increased risk of diseases of the nervous system overall (RR 9.66, 95% CI 7.24-12.89) (Table 2). This is inclusive of the CNS findings of dystonia, extrapyramidal and movement disorders, and epilepsy or recurrent seizures. Due to small patient numbers, we were unable to calculate the true prevalence and relative risk of several nervous system diagnoses, including multiple sclerosis, diseases of the myoneural junction, and inflammatory CNS disease. A list of additional diagnoses analyzed but not significantly different between cohorts or with indeterminate prevalence is available in Supplementary Table 1.
Prevalence of selected nervous system diagnoses in populations with and without Treacher Collins Syndrome (TCS) after propensity matching for age, race, and ethnicity.

*TriNetX rounds patient counts from 1-9 to 10 to maintain anonymity. As a result, we report the minimum possible relative risk and 95% CI.
Patients with TCS also had an increased risk of overall congenital malformations of the circulatory system (RR 8.5, 95% CI 4.44-16.28) (Table 3). The greatest risk was associated with atrial septal defects (RR 4.2, 95% 2.12-8.33). Too few patients were available after propensity match in our control cohort to determine the true prevalence for ventricular septal defect and patent ductus arteriosus, among other conditions.
Prevalence of selected cardiovascular diagnoses in populations with and without Treacher Collins Syndrome (TCS) after propensity matching for age, race, and ethnicity.

*TriNetX rounds patient counts from 1-9 to 10 to maintain anonymity. As a result, we report the minimum possible relative risk and 95% CI.
Table 4 contains our findings of otologic complications. Patients with TCS had an increased risk for eustachian tube dysfunction, affecting 9.79% of the population (RR ≥ 10.9, 95% CI 5.73-20.72). Prevalence of otitis media was also elevated in the TCS group with a Relative Risk of 13 (95% CI 6.87-24.60). Nearly 40% of our patients with TCS had a diagnosis of conductive hearing loss, 44 times the prevalence of the condition in the control cohort (95% CI 24-83).
Prevalence of selected otologic diagnoses in populations with and without Treacher Collins Syndrome (TCS) after propensity matching for age, race, and ethnicity.

*TriNetX rounds patient counts from 1-9 to 10 to maintain anonymity. As a result, we report the minimum possible relative risk and 95% CI.
Discussion
Nervous System
The pathophysiology linking TCS and diseases of the nervous system is unknown. However, recent genotyping has revealed a gene mutated in some cases of TCS that overlaps with Central Nervous System degeneration. In 2015, Nature Communications published the discovery of a novel RNA polymerase III mutation associated with a class of neurodegenerative disorders called demyelinating leukodystrophy. This novel mutation involved POLR1C, the gene also responsible for causing autosomal-recessive TCS. In the case of POLR1C demyelinating leukodystrophy, patients may experience progressive ataxia, cerebellar atrophy and supratentorial hypomyelination, as well as tonic-clonic seizures. Imaging has revealed hypointensity of the thalamus, internal capsule, and substantia nigra.8 A 2019 study included 23 individuals recruited based on their diagnosis of POLR1C-variant RNA Polymerase III-related leukodystrophy. Dystonia was present in 32% of the patients, and the 22 that survived beyond infancy showed cerebellar signs. On MRI, all 22 showed diffuse hypomyelination; 45% had hypomyelination of the internal capsule and cerebellar dentate nucleus. Five had features of abnormal craniofacial development, with one reportedly having “clear” TCS features.9
Notably, the POLR1C gene encodes for two subunits, and the mechanisms by which a mutation causes TCS and leukodystrophy are different. Leukodystrophy-inducing mutations affect one subunit but not the other, whereas the TCS mutation leads to normal assembly of both, but impairs the protein’s targeting of the nucleolus.10 However, it is possible that there is overlap in the phenotype of these different mutations of the same gene, resulting in the increased prevalence of neurodegenerative disorders seen in individuals with TCS.
It is theorized that POLR1C mutations account for approximately 1.2% of TCS cases.3 If we apply that estimate to our 1,114 included patients, we would expect around 13 of them to have the POLR1C mutation. Interestingly, our sample included 15 patients with dystonia and even more with epilepsy or recurrent seizures, at 42. These numbers may reflect patients who have received more than one of the diagnoses. Although it is impossible to say which TCS mutation the individuals harbored, these findings do suggest a need for future research into the genetic basis for CNS anomalies in patients with TCS.
Cardiovascular System
Cardiovascular anomalies in TCS are more often discussed than neurodegenerative diseases; however, our sample size of 1,114 patients is far larger than that of any existing study on the subject. A 2018 systematic review of 15 studies found 8 cardiovascular malformations among 144 included patients, an overall prevalence of 5.6%.11 All eight were in patients with TCOF1 gene mutations. A 2021 study of 248 patients found that cardiac anomalies made up 14% of the extracraniofacial anomalies diagnosed within the cohort, totaling 13 cases with a prevalence of 5.2%.12 Another study of 146 patients found a higher prevalence of cardiac malformations, which affected 11.7% of their TCOF1 cohort and 10.8% of TCS patients overall,13 consistent with our finding of approximately 11%. The specific diagnoses most commonly determined in these studies were patent ductus arteriosus and ventricular and atrial septal defects. We found atrial septal defect to be disproportionately elevated within our cohort, with the prevalence of ventricular septal defect likely increased as well with a relative risk and confidence interval of at least 1.8 (95% CI 0.84-3.88).
The TCOF1 gene is involved in mRNA formation in neural crest cells which are a major component of the pharyngeal arches. Mutations in TCOF1 are predicted to deplete the number of cranial neural crest cells migrating to the first and second pharyngeal arches that give rise to structures of the head and face.14 However, it is possible that the neural crest cells influenced by TCOF1 are not limited to these two pharyngeal arches and populate developing cardiac structures as well.12 Little research has been done into the pathophysiology behind TCS and cardiovascular anomalies, perhaps due to their rarity. However, our analysis indicates that cardiac anomalies in TCS are somewhat more common than previous, smaller studies have calculated. While some other figures have hovered around 5% for cardiac and cardiovascular malformations,11,12 we found an overall prevalence of congenital malformations of the circulatory system to be nearly 8%.
Otologic Complications
As TCS affects the bones of the head and face, often including the development of the structures of the outer, middle, and inner ear, conductive hearing loss is a known and expected complication of TCS. However, previous studies have reported varying prevalence of conductive hearing loss, from 40% to over 90% commonly reported.3,15 In our large cohort, conductive hearing loss affecting approximately 40% of patients. Furthermore, the frequency of eustachian tube dysfunction in patients with TCS has not previously been reported, despite affecting nearly 10% of our cohort. We also found that TCS increased the risk of diagnosed otitis media 13 times in our propensity matched cohort. This increased risk may be mediated by eustachian tube dysfunction: when obstructed, negative pressure is able to build in the middle ear, allowing nasopharyngeal bacteria to backflow into this space.16 Prior to propensity matching, our findings of acute otitis media may also be influenced by the significantly younger age of our TCS cohort, predisposing them to such infections and potentially inflating our numbers. However, we accounted for age in our propensity match to eliminate the effects of age. It is important to recognize any possibility of an increased risk otitis media as the infection can precipitate conductive hearing loss. Given that TCS patients are already at an increased risk for hearing loss due to developmental abnormalities, it is necessary to consider the risk for otitis media to promote prompt treatment and prevention of further auditory damage.
Limitations
This population-level study is not without its limitations. The EHR aggregation platform used is dependent upon conditions being recognized, diagnosed, and documented in the underlying EHRs of contributing healthcare systems. As previously discussed, it is likely that some individuals with TCS who have mild craniofacial abnormalities go undiagnosed, which would actually lead to understating the TCS associated risk profiles described here. Alternatively, several relative risks could not be calculated due to TriNetX rounding the number of patients with any diagnosis from 1-9 up to 10. While this preserves anonymity, it inhibits analysis of true prevalence and may lead to underreporting of increased risk. We reported conditions for which a relative risk could not accurately be calculated within our Tables, including Supplementary Table 1.
Potentially contributing to overestimation, individuals with TCS may be more likely to undergo a thorough newborn screen or interact more frequently with the healthcare system, leading to increased diagnosis and documentation of other health conditions. Although we attempted to randomly reduce our control cohort in order to perform a propensity match, due to the inability of TriNetX to propensity match such a large number of patients, our requirement of an ambulatory visit within three months of our search for patients without TCS may select for patients with more health conditions or more interaction with the healthcare system. However, we would expect any effect of this to underestimate the relative risk of TCS.
Finally, we only have access to the TCS diagnosis in the TriNetX platform, but not the specific mutation responsible for each case, limiting our ability to hypothesize the biological mechanisms underlying our proposed associated conditions. As such, other co-occurring genetic syndromes may be responsible for the observed phenotypes; molecular genetics data would be required to definitively link TCS and our observed abnormalities. Similarly, as our diagnosis is limited to the ICD-10 code for mandibulofacial dysostosis, although most commonly associated with TCS,7 it is possible that our cohort also includes similar-appearing but far more infrequent dysostoses affecting the face. One such example is Nager syndrome, a condition with both craniofacial and limb abnormalities.17
Conclusion
Little is known about the epidemiology or biological basis for extracraniofacial anomalies associated with TCS. Minimal prior data exists on an association between TCS and disorders of the central nervous system, although we found significantly increased risk in patients with TCS of co-occurring dystonia and epilepsy or recurrent seizures. Although smaller studies have postulated a connection between TCS and congenital cardiovascular deformities, we believe their numbers may underestimate a true relationship between the conditions and recommend thorough screening of all patients with diagnosed or suspected TCS. Finally, recognizing the high prevalence of conductive hearing loss in individuals with TCS, as well as the risk for otitis media, we encourage a heightened awareness for the additional long-term damage that recurrent infections may have on auditory function. This study also demonstrates the increasing potential and value for large, aggregated EHR platform to uncover associations with rare conditions that previously could not be well described. Future research on TCS should determine the specific causal mutation of the included patients, as well as any extracraniofacial malformations noted for each.
Research Ethics and Patient Consent
Patient consent is not applicable and is not needed. The aggregated data used in this manuscript only contained de-identified data and has been deemed exempt from the Western Institutional Review Board (IRB) by a qualified expert as defined in Section §164.514(b)(1) of the HIPAA Privacy Rule.
Supplemental Material
sj-docx-1-cpc-10.1177_10556656231187302 - Supplemental material for Treacher Collins Syndrome Associated with Disproportionate Nervous System, Cardiovascular, Otologic Complications Among 1,114 Patients
Supplemental material, sj-docx-1-cpc-10.1177_10556656231187302 for Treacher Collins Syndrome Associated with Disproportionate Nervous System, Cardiovascular, Otologic Complications Among 1,114 Patients by Jacqueline Kloos and David C. Kaelber in The Cleft Palate Craniofacial Journal
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.