
Abstract
Objective
Auriculocondylar syndrome (ARCND) is a set of rare craniofacial malformations characterized by variable micrognathia, ear malformations, and mandibular condyle hypoplasia, and other accompanying features with phenotypic complexity. ARCND2 caused by pathogenic variants in the PLCB4 gene is a very rare disease with less than 50 patients reported and only 36 different variants of the PLCB4 gene recorded in HGMD. This study aims to enrich the patient resources, clinical data and mutational spectrum of ARCND2.
Design
Case series study.
Setting
Guangzhou Women and Children's Medical Center and Guangdong Women and Children Hospital.
Patients
Two Chinese patients with ARCND2.
Main Outcome Measures
Clinical, radiological and molecular findings.
Results
Both the two patients presented with craniofacial and ear malformations, and feeding difficulties. Whole exome sequencing identified two different variants of the PLCB4 gene in these two patients with a heterozygous allele and a de novo mode of inheritance respectively. Patient 1 carried a known pathogenic c.1861C > T(p.Arg621Cys) missense variant, whereas Patient 2 had a novel c.225 + 1G > A splicing variant. Sanger sequencing confirmed the presence of PLCB4 variants in the proband and absence in the unaffected parents. These two PLCB4 variants were suggested as disease-causing candidates for these two patients. During a 5-year follow-up, Patient 2 gradually manifested crowded teeth, underweight, motor delay and intellectual disability.
Conclusions
In this study, we report two Chinese patients with ARCND2, describe their clinical and mutational features, and share a 5-year follow-up of one patient. Our study adds two additional patients to ARCND2, reveals a novel PLCB4 variant, and expands the phenotypic and genotypic spectrum.
Keywords
Introduction
Auriculocondylar syndrome (ARCND), also known as “question-mark ear syndrome”, was first described in 1970. 1 It is a set of rare craniofacial malformations involving the first and second pharyngeal arches with an estimated prevalence of less than 1 per 1,000,000 people.2,3 The typical clinical features of ARCND include variable micrognathia, ear malformations, and mandibular condyle hypoplasia accompanied by other phenotypes of prominent cheeks, microstomia, glossoptosis, cleft palate, facial asymmetry, crowded teeth, and hearing impairment.3–5 However, ARCND is still not well-known because of high inter- and intra-familial phenotypic variability.5,6
In ARCND, three different genetic subtypes have been identified, that is ARCND1 (OMIM # 602483), ARCND2 (OMIM # 614669), and ARCND3 (OMIM # 615706) caused by pathogenic variants in the GNAI3, PLCB4 and EDN1 gene respectively.7,8 Moreover, a new locus covering most of the HDAC9 gene with a telomeric location of the TWIST1 gene is suggested as a candidate cause of ARCND recently. 9
Although ARCND2 is the most frequent form accounting for 58% of the patients with ARCND,3,9 less than 50 patients have been reported to have ARCND2 worldwide.3,4,6,7,10–18 To date, only 36 different variants of the PLCB4 gene have been documented in the Human Gene Mutation Database (HGMD, Professional 2023.4). Among them, 23 variants are disease-causing, and 12 variants are possible disease-causing.
In the present study, we described two new Chinese patients with ARCND2, analyzed their clinical, radiographic and genetic findings, and provided a 5-year follow-up of one patient.
Materials and Methods
Patients
Patient 1 was enrolled in Guangzhou Women and Children's Medical Center, and Patient 2 was enrolled in Guangdong Women and Children Hospital. All subjects were southern Chinese and of Han ethnicity. All parents were unaffected with normal appearances.
Clinical Information
The clinical data were collected and evaluated by clinicians. Physical examinations were performed by physicians. Family history was inquired by genetic counselors. Routine biochemical, metabolic and chromosomal analyses were detected in the hospital's clinical laboratory center. Brain magnetic resonance imaging (MRI), craniofacial computed tomography (CT) and oral X-ray were carried out in the hospital's medical imaging center. Electroencephalogram (EEG) and brainstem auditory evoked potential (BEAP) were performed by neurologists. Electronic laryngoscopy was performed by otolaryngologists.
Molecular Analyses
Genomic DNA (gDNA) was extracted from whole blood samples using DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany). As previously described,19,20 whole exome sequencing (WES) was performed according to the manufacturers’ protocol using Agilent SureSelect Human All Exon V6 kit (Agilent, Santa Clara, United States) and Illumina HiSeq X Ten platform (Illumina, San Diego, United States) with a depth over 100X and coverage over 99%.
The acquired data were processed upon an established analysis pipeline for variant calling and functional annotation to identify the disease-causing variants. Firstly, the raw reads were evaluated using fastp, BWA, SAMtools and Sambamba software. Secondly, the SNVs, InDels and CNVs were called using SAMtools, BCFtools, ANNOVAR and CoNIFER software. Thirdly, the variants were annotated using different databases or software. The frequency was acquired using SNP databases, including 1000Genomes, ESP6500, ExAC, GnomAD and NovoDb, a local population's database; the pathogenicity was annotated by mutation databases, including HGMD and ClinVar, or predicted by dbscSNV, SPIDEX, Interpro, SIFT, PolyPhen-2, MutationTaster, LRT, MutationAssessor, FATHMM, PhyloP, SiPhy, CADD, GERP++, MCAP and REVEL software; the gene-disease correlation was provided by OMIM, KEGG and GO databases; the references were supplied by PubMed database.
Subsequently, the polymorphic alleles with a frequency over 1% in any of SNP databases were excluded. The proband-parents trio was analyzed with family model to screen de novo, homozygous and compound heterozygous variants respectively. The causative genes of ARCND, including GNAI3, PLCB4, EDN1, HDAC9 and TWIST1, were further reviewed.
For confirmatory Sanger sequencing, the exon sequences together with adjoining intron boundaries of the PLCB4 gene (NG_032790.2, NM_000933.4) were amplified and sequenced using an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, United States). The known pathogenic missense variant was confirmed by HGMD and ClinVar, while the functional consequences of the novel splicing variant was predicted by in-silico analyses using MutationTaster, FATHMM, NetGene2, NNSPLICE 0.9, and RNA Splicer software. The pathogenicity of variants was evaluated according to the American College of Medical Genetics (ACMG) guidelines. 21
Follow-up
Patient 1 was lost to follow-up at a half of year after diagnosis, whereas Patient 2 kept irregular clinic visits with an interval of 3–14 months. Height and weight of Patient 2 were measured at every visit.
Results
Clinical Presentations
Patient 1
Patient 1 was the first live child of the non-consanguineous healthy parents, who had a previous induced termination of pregnancy. She was born naturally at 41 weeks of gestation. There was no history of perinatal asphyxia. After birth, she was found to have prominent cheeks, mild micrognathia, microstomia, low-set ears, and bilateral malformed auricles with post-auricular skin tags and absence of ear lobes (Figure 1A). She did not pass the newborn hearing screening. Subsequently, she gradually presented with feeding difficulties, growth retardation, hearing impairment, and hypotonia.
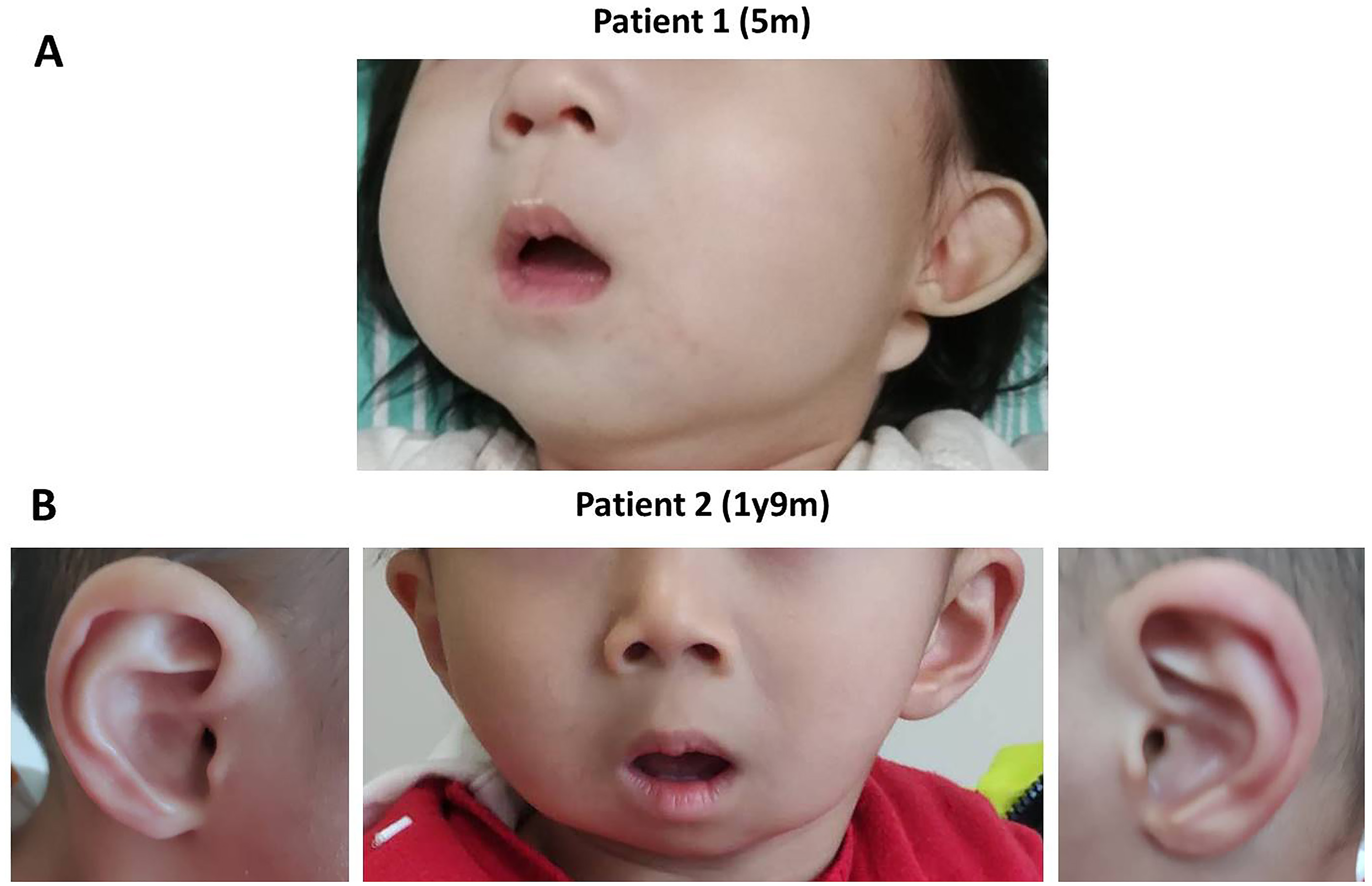
The photos of the two ARCND2 patients.
She was 5 months old when she first visited our clinic because of diarrhea and seizure, and presented low body weight of 5.4 kg which is below −2 standard deviation (SD) of the reference range. 22 She could babble and hold head up but with poor head control. No obvious abnormality was found in her blood and urine biochemical and metabolic profiles, brain MRI, and EEG. BEAP showed impaired bilateral conduction auditory pathway with increased auditory threshold. Her karyotype and chromosomal microarray analysis (CMA) were normal.
Patient 2
Patient 2 was the fifth child of the consanguineous healthy parents, who already had four healthy daughters. He was born naturally at 42 weeks of gestation. There was no history of birth asphyxia, and he passed the newborn hearing screening. After birth, he manifested prominent cheeks, micrognathia, mandibular hypoplasia, microstomia, and malformed helices (Figure 1B). More seriously, he suffered from opisthotonus, apneas and dysphagia, and was admitted to the neonatal intensive care unit for a week of nasal feeding, swallowing function training, and continuous positive airways pressure.
He was 1 month old when he first visited our clinic with normal height and weight (Figure 2). No obvious abnormality was found in his blood and urine biochemical and metabolic profiles, brain MRI, and BEAP. His karyotype and CMA were normal. Electronic laryngoscopy showed curly epiglottis with the aryepiglottic folds closer to glottis.
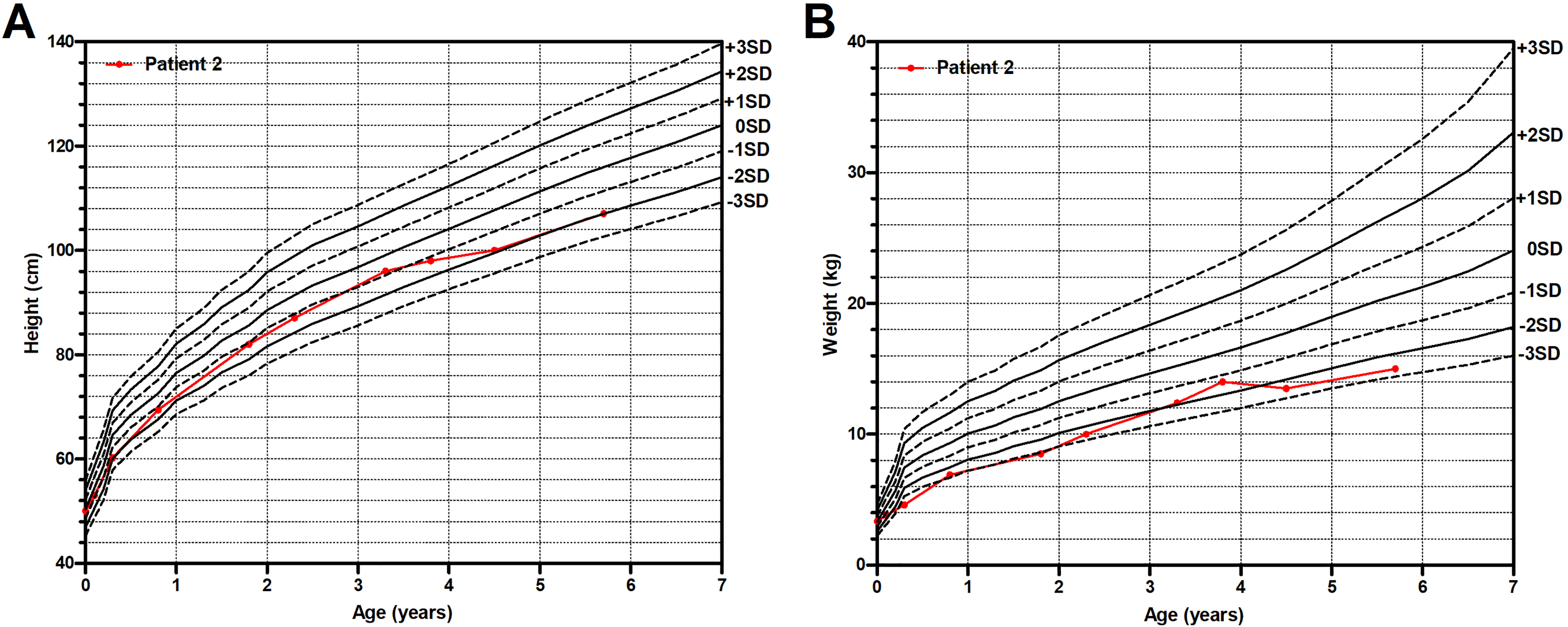
The growth curve of Patient 2.
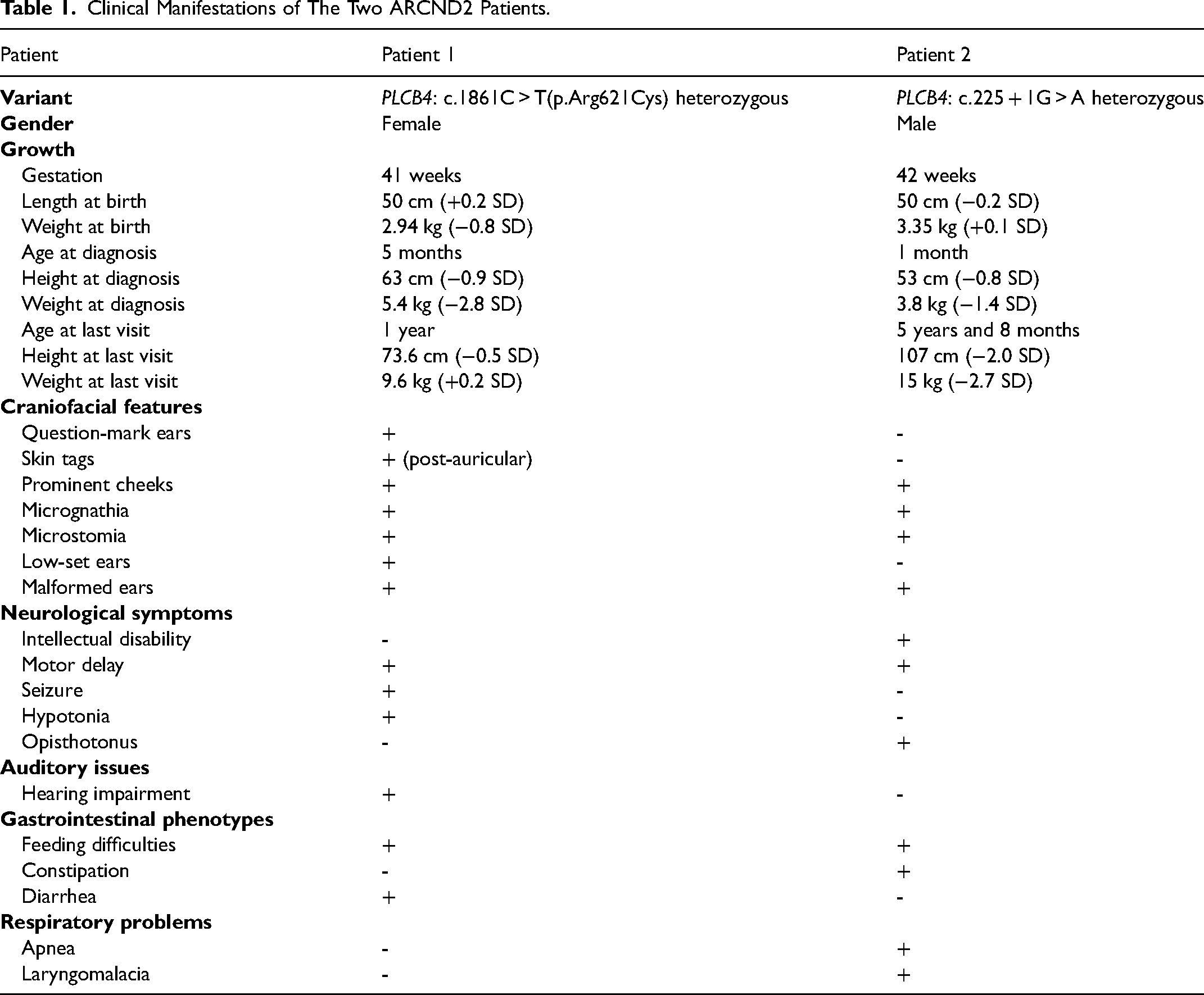
Based on the facial appearance, growth and development of these two patients (Table 1), both were suspected of having a syndrome. However, due to their young age, limited observations and examinations could not determine the specific disease.
Clinical Manifestations of The Two ARCND2 Patients.
Molecular Analyses
To investigate the phenotype-producing variants, WES of the proband-parents trio was performed. Two different variants in the PLCB4 gene were identified in these two patients with a heterozygous allele and a de novo mode of inheritance respectively. Patient 1 carried a known pathogenic c.1861C > T(p.Arg621Cys) missense variant,7,13,14 whereas the c.225 + 1G > A variant in Patient 2 is a novel one not found in SNP or mutation databases. Located in a canonical donor splice site, this c.225 + 1G > A variant putatively disrupted the normal splice site (Figure S1). It was predicted to be deleterious by in-silico analyses and classified as likely pathogenic variant according to the ACMG guidelines (Table S1). No other suspicious variants were found in terms of the inheritance pattern and clinical manifestations. Only the variants in the previously identified causative gene for ARCND2 were further considered.
Sanger sequencing subsequently confirmed the presence of PLCB4 variants in the proband and absence in the unaffected parents (Figure 3). On account of clinical and molecular findings, these two PLCB4 variants were suggested as disease-causing candidates for these two patients.
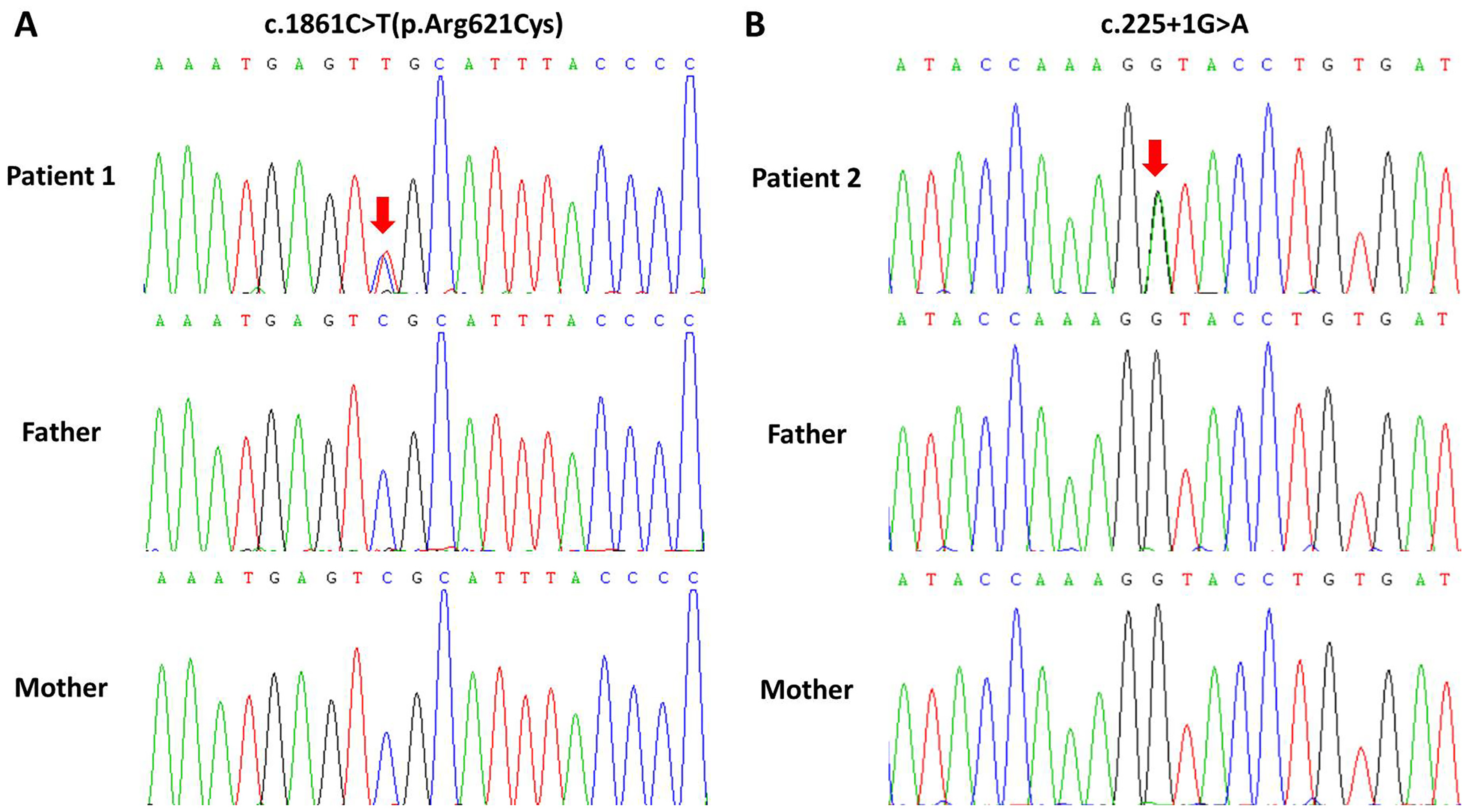
Sanger sequencing chromatograms of the enrolled family. The red arrows indicate the variant site.
Follow-up of Patient 2
Only Patient 2 had a follow-up lasting for 5 years with an interval of 3–14 months, whereas Patient 1 was lost to follow-up at a half of year after diagnosis with only one-time physical examination at 1 year old.
During follow-up, feeding problems of Patient 2 gradually became prominent with weak chewing ability and poor tongue flexibility. Oral X-ray showed crowded teeth (Figure S2), and craniofacial CT with 3-D reconstruction revealed bilaterally foreshortened vertical mandibular rami and absence of a condylar fossa at 2 years old (Figure S3). Compared with that at 2 days old, his craniofacial CT showed little change on mandibular angle but a much smaller tongue base-oropharyngeal space and a slightly narrowed sella-nasion-point B (SNB) angle at 2 years old and 3 years old (Figure 4).
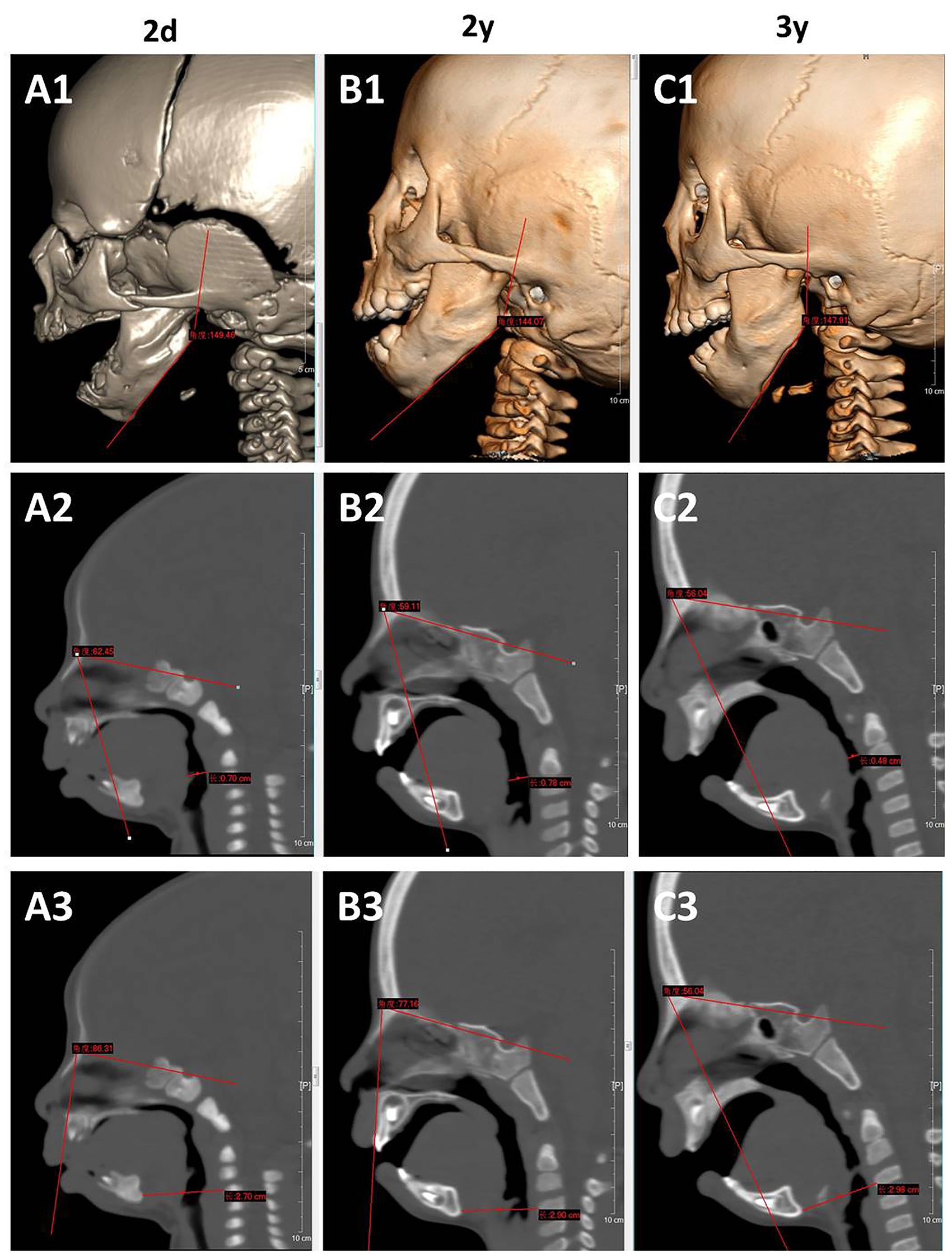
Comparison of craniofacial CT of Patient 2 at 2 days old, 2 years old and 3 years old.
Until 5 years and 8 months old, Patient 2 maintained a normal height above −2 SD (Figure 2A). He was underweight from 4 months old to 2 years and 4 months old because of liquid diet alone and had a catch-up afterwards by adding solid foods, whereas his weight still fell outside the reference range after 4 years and 6 months old (Figure 2B). He had mild to moderate constipation with the need of stool softeners occasionally.
Moreover, Patient 2 showed delayed motor milestones as he achieved head control at 4 months old, stable sitting at 1 year old, and independent walking at 2 years old. His developmental quotient of Gesell Developmental Schedules was 57.6 at 2 years old. 23 He also had a mild defect in language development due to small mandible as his comprehension and expression skills at 2 years old were equivalent to the level of a 1.5-year-old child.
Discussion
ARCND is characterized by the triad of core features, micrognathia, ear malformations, and mandibular condyle hypoplasia, and other accompanying phenotypes of prominent cheeks, microstomia, glossoptosis, cleft palate, facial asymmetry, crowded teeth, and hearing impairment.3–5 ARCND2 caused by deleterious variants of the PLCB4 gene is a very rare disease with less than 50 patients worldwide.3,4,6,7,10–18
ARCND2 presents genetically heterogeneity with typical autosomal dominant or rare autosomal recessive inheritance model.3,16,18 Previous studies showed that most of autosomal dominant ARCND2 cases are resulted from heterozygous missense variants falling in the catalytic domains of the PLCB4 gene with a predicted role of a dominant-negative effect,7,13,16 which is proven by Kanai et al. with in vitro and in vivo evidences. 24 In contrast, homozygous loss-of-function (LoF) variants in the PLCB4 gene lead to autosomal recessive ARCND2, whereas heterozygous carriers of LoF variants are asymptomatic.4,13,16
However, not all cases fit this observation well. Fan et al. found a homozygous c.991T > C(p.Y331H) missense variant in the PLCB4 gene could cause ARCND2 with an autosomal recessive trait, 17 which is further supported by the ARCND2 case recently reported by El Fizazi et al. with a homozygous PLCB4 c.2050G > A(p.Gly684Arg) missense variant, 18 suggesting a variant moving further away from the catalytic core might be hypomorphic and only causes phenotype in a homozygous manner.
In our study, Patient 1 shared the classic phenotypes of ARCND (Figure 1 and Table 1) and harbored a known pathogenic PLCB4 c.1861C > T(p.Arg621Cys) missense variant with a heterozygous allele,7,13,14 leading to a definite diagnosis of autosomal dominant ARCND2. Patient 2 presented with mandibular condyle hypoplasia, micrognathia, ear malformations, prominent cheeks, microstomia, and crowded teeth of ARCND, though he did not have the symbolic question-mark ears (Table 1, Figure 1, Figure 4, Figure S2 and Figure S3). WES of the proband-parents trio suggested a heterozygous c.225 + 1G > A LoF variant of the PLCB4 gene as a candidate disease-cause for Patient 2, and no other suspicious variants were found. On account of clinical and molecular findings, a possible diagnosis of autosomal dominant ARCND2 was made for Patient 2.
Li et al. recently identified a heterozygous PLCB4 LoF variant, c.2980delA(p.Met994*), in a Chinese boy with short stature, facial asymmetry, and development delay, 25 which supports our supposition that monoallelic LoF variant in the PLCB4 gene could lead to autosomal dominant ARCND2. Furthermore, three of five predictive scores (LOEUF, sHet and pHaplo) in DECIPHER database suggest that PLCB4 gene is likely to be dosage-sensitive. As incomplete penetrance and/or variable expression within families of autosomal dominant ARCND2 are observed and much fewer autosomal recessive ARCND2 patients are recognized, 13 it is probable that incomplete penetrance occurs in heterozygous carriers of PLCB4 LoF variants.
To date, only 35 different disease-causing or possible disease-causing variants of the PLCB4 gene have been documented in HGMD (Professional 2023.4) to cause ARCND2 or related phenotypes. In this study, two different variants of the PLCB4 gene were identified. Among them, c.225 + 1G > A splicing variant in Patient 2 is novel, which expands the mutational spectrum of the PLCB4 gene.
In addition, Vegas et al. recently summarized that 44% (12/27) ARCND patients developed psychomotor delay. 13 In our study, Patient 1 presented motor delay, while Patient 2 was affected by motor delay and intellectual disability. The findings of our patients support their suggestion that neurodevelopmental disorder should be listed as additional feature associated with ARCND.
Conclusions
Our study presents two novel Chinese patients of ARCND2, provides their clinical and molecular features, and shares a 5-year follow-up of one patient, which enriches the patient resources and clinical data. Our study also adds a new likely pathogenic PLCB4 splicing variant to ARCND2, which expands the mutational spectrum of this disease.
Supplemental Material
sj-docx-2-cpc-10.1177_10556656241234575 - Supplemental material for Two Chinese Patients of Auriculocondylar Syndrome 2: A Novel PLCB4 Splicing Variant and 5-Year Follow-up
Supplemental material, sj-docx-2-cpc-10.1177_10556656241234575 for Two Chinese Patients of Auriculocondylar Syndrome 2: A Novel PLCB4 Splicing Variant and 5-Year Follow-up by Yunting Lin, Ye Zhang, Jian Ma, Shu Liu, Yongxi Liu, Chaoxiang Yang, Chunhua Zeng and Xianqiong Luo in The Cleft Palate Craniofacial Journal
Supplemental Material
sj-pdf-1-cpc-10.1177_10556656241234575 - Supplemental material for Two Chinese Patients of Auriculocondylar Syndrome 2: A Novel PLCB4 Splicing Variant and 5-Year Follow-up
Supplemental material, sj-pdf-1-cpc-10.1177_10556656241234575 for Two Chinese Patients of Auriculocondylar Syndrome 2: A Novel PLCB4 Splicing Variant and 5-Year Follow-up by Yunting Lin, Ye Zhang, Jian Ma, Shu Liu, Yongxi Liu, Chaoxiang Yang, Chunhua Zeng and Xianqiong Luo in The Cleft Palate Craniofacial Journal
Footnotes
Acknowledgements
The authors would like to thank the enrolled subjects for participation in this study. The authors thank the hospital's medical detection departments for assistance with imaging and biochemical examinations. The authors also thank Helmut Grasberger and Guoqing Hou from the University of Michigan Medical School for language correction.
Author Contributions
All the listed authors were involved in drafting or editing this article, and approved its submission and publishment. XL and CZ designed the study. XL, CZ, JM, SL, YXL and CY enrolled the family and collected the clinical data. YZ, YTL and JM performed the experiments and analyzed the data. YTL, YZ and CZ wrote the paper.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by Research Fund of Guangzhou Science and Technology Innovation Commission (grant number 202102080455 and 202102020133) and Joint Research Fund of Guangdong Basic and Applied Basic Research Commission (grant number 2021A1515220168).
Ethics Approval and Consent
This study was approved by the Institutional Review Board of Guangzhou Women and Children's Medical Center (Guangzhou, China) (No. 2015-112) and the Institutional Review Board of Guangdong Women and Children's Hospital (No. 2018010045). Informed consents were obtained from the participants or the guardians of under-aged participants.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.