
Abstract
Objective
To describe the frequency and types of hearing loss in children with syndromic and non-syndromic craniosynostosis.
Design
Retrospective cohort study.
Setting
Large tertiary pediatric hospital.
Patients
Children with craniosynostosis that underwent at least one audiological evaluation between the years of 2010 and 2021 at a single institution. Hearing loss was defined as conductive or permanent (sensorineural or mixed).
Results
Of 130 total children examined, 107 (82.3%) had non-syndromic craniosynostosis and 23 (17.7%) had syndromic craniosynostosis. Within the non-syndromic cohort, 77 (72%) had normal hearing and 30 (28%) had hearing loss, of which 21/30 (70%) had conductive hearing loss and 9/30 (30%) had permanent hearing loss. Within the syndromic cohort, two (9.5%) had normal hearing and 21 (90.5%) had hearing loss, of which 16/21 (76.2%) had conductive hearing loss and 5/21 (23.8%) had permanent hearing loss. Multivariable analysis involving syndromic status, anatomical type of craniosynostosis, and medical complexity revealed that patients with syndromic craniosynostosis had higher odds for conductive and permanent hearing loss (49.80 OR, P = .002 and 34.91 OR, P < .05, respectively). Patients with unicoronal craniosynostosis and those with significant medical comorbidities had higher odds for permanent hearing loss (19.50 OR, P = .03 and 23.36 OR, P < .05, respectively).
Conclusions
Children with craniosynostosis had high rates of hearing loss, especially those with syndromic or unicoronal craniosynostosis and those with significant medical comorbidities. Twenty-eight percent of patients with non-syndromic craniosynostosis also had hearing loss. Larger prospective studies are necessary to more precisely estimate hearing loss associated with non-syndromic craniosynostosis.
Introduction
Craniosynostosis is a congenital condition defined as the premature fusion of one or more cranial sutures 1 that can present in isolation or in the context of a syndrome. 2 Isolated or non-syndromic craniosynostosis is estimated to affect 5.2 in 10 000 live births, globally, and is more common than syndromic presentations. 3 Complications due to craniosynostosis may include poor head shape, ocular disorders, increased intracranial pressure, 4 and neurodevelopmental disorders.5–7 Hearing loss is another possible complication, 8 though this is not fully described in the current literature. The definitive treatment of craniosynostosis is surgical intervention to release the affected cranial sutures, promoting normalization of head growth, reducing intracranial pressure, and potentially reducing negative developmental outcomes.9,10
Hearing loss is often categorized into three types: conductive hearing loss (CHL) due to a disruption of the auditory signal in the outer or middle ear, sensorineural hearing loss (SNHL) due to abnormalities in the cochlea or auditory nerve, or mixed hearing loss (MHL) which is a combination of CHL and SNHL. Otitis media with effusion (OME), an excess of fluid in the middle ear, is a common cause of CHL in children, with most children experiencing OME at least once by the age of two. 11 While most episodes of OME resolve spontaneously, a subset of children have persistent episodes that continue to impact hearing for a year or longer. 12 Hearing loss has been associated with poor speech and language development in children 13 thus early assessment and intervention is recommended. 14
There are over 180 syndromes associated with craniosynostosis with some of the most common including Muenke syndrome, Crouzon syndrome, Apert syndrome, and Pfeiffer syndrome. 2 Hearing loss has been documented in children with many presentations of syndromic craniosynostosis.15,16 Potential hypotheses for the association between hearing loss and a syndrome include genetic or anatomic causes, or some combination of both. 17 SNHL is often a hallmark finding in Muenke syndrome, and 70% of individuals present with some form of hearing loss. 18 An animal model of Muenke syndrome has shown abnormal development of inner ear cells that are likely the cause of the SNHL. 19 While other presentations of syndromic craniosynostosis may also have a SNHL component, CHL is more commonly seen. 20 CHL can be attributed to middle ear and palatal anomalies that may result in OME or other disruptions in the auditory signal. 21 Agochukwu et al found that, in a review across multiple studies in the literature, hearing loss occurs in 61% of individuals with Muenke syndrome, 80% of individuals with Apert syndrome, 92% of individuals with Pfeiffer syndrome, 74% of individuals with Crouzon syndrome, and 68% of individuals with Jackson-Weiss syndrome. 21
Conversely, hearing outcomes in non-syndromic craniosynostosis are not as well documented. One study found that, of 113 children with non-syndromic craniosynostosis, 9% of children had hearing loss (7% CHL and 2% SNHL) but that 36% of the children from that same cohort had OME. 22 In another study comparing audiologic evaluations of preschool-aged children with syndromic and non-syndromic craniosynostoses, investigators found that 82% (18/22) of children with syndromic craniosynostosis had hearing loss compared with 22% (2/9) of children with non-syndromic craniosynostosis. 23 Looking exclusively at non-syndromic sagittal craniosynostosis, a different study found that 26% (15/57) of children had some degree of hearing loss. 24 Overall, there is a paucity of studies on hearing loss in non-syndromic craniosynostosis compared with those in syndromic craniosynostosis.
The aim of this study is to better describe types and frequencies of hearing loss in children with syndromic and non-syndromic craniosynostosis. To achieve this, the most common causes of hearing loss in the pediatric population had to be distinguished from hearing loss due to craniosynostosis. Thus, a multivariable model was developed that examines whether three factors were associated with conductive or permanent hearing loss: syndromic status, anatomical type of craniosynostosis, and medical complexity. Our hypothesis is that genetic syndromes associated with craniosynostosis will be the primary predictor for hearing loss, especially permanent hearing loss, and craniosynostosis itself may predispose patients to higher rates of conductive or permanent hearing loss.
Methods
From an IRB approved retrospective cohort study, electronic medical records were reviewed from a single, large, tertiary pediatric hospital with a craniofacial team approved by the American Cleft Palate Craniofacial Association (ACPA). The following inclusion criteria was utilized: children with a diagnosis of craniosynostosis that had surgical treatment of craniosynostosis between January 1st, 2010, and December 31st, 2021. A total of 370 children met initial inclusion criteria; a subset of 149 children (38.5% of the original 370) had at least one documented audiological evaluation within this timeframe, beyond the initial newborn hearing screen (Figure 1). However, since younger patients toward the end of the study window may not have had the same opportunity to develop hearing loss or be referred for audiological testing, those with normal hearing who were less than 36 months old at the end of this timeframe were excluded from the normal hearing group, reducing the sample size to 130 children (Figure 1). Based on clinical experience, this was considered sufficient time for referral to audiology for the most common causes of hearing loss in the general pediatric population. A sensitivity analysis was also performed (n = 88 patients), repeating the analyses with stricter inclusion criteria for the normal hearing group: the latest available hearing screen must have occurred when the patient was at least 36 months old (Figure 1). This was done to further ensure that all patients had an equal opportunity to be referred and assessed for hearing loss.
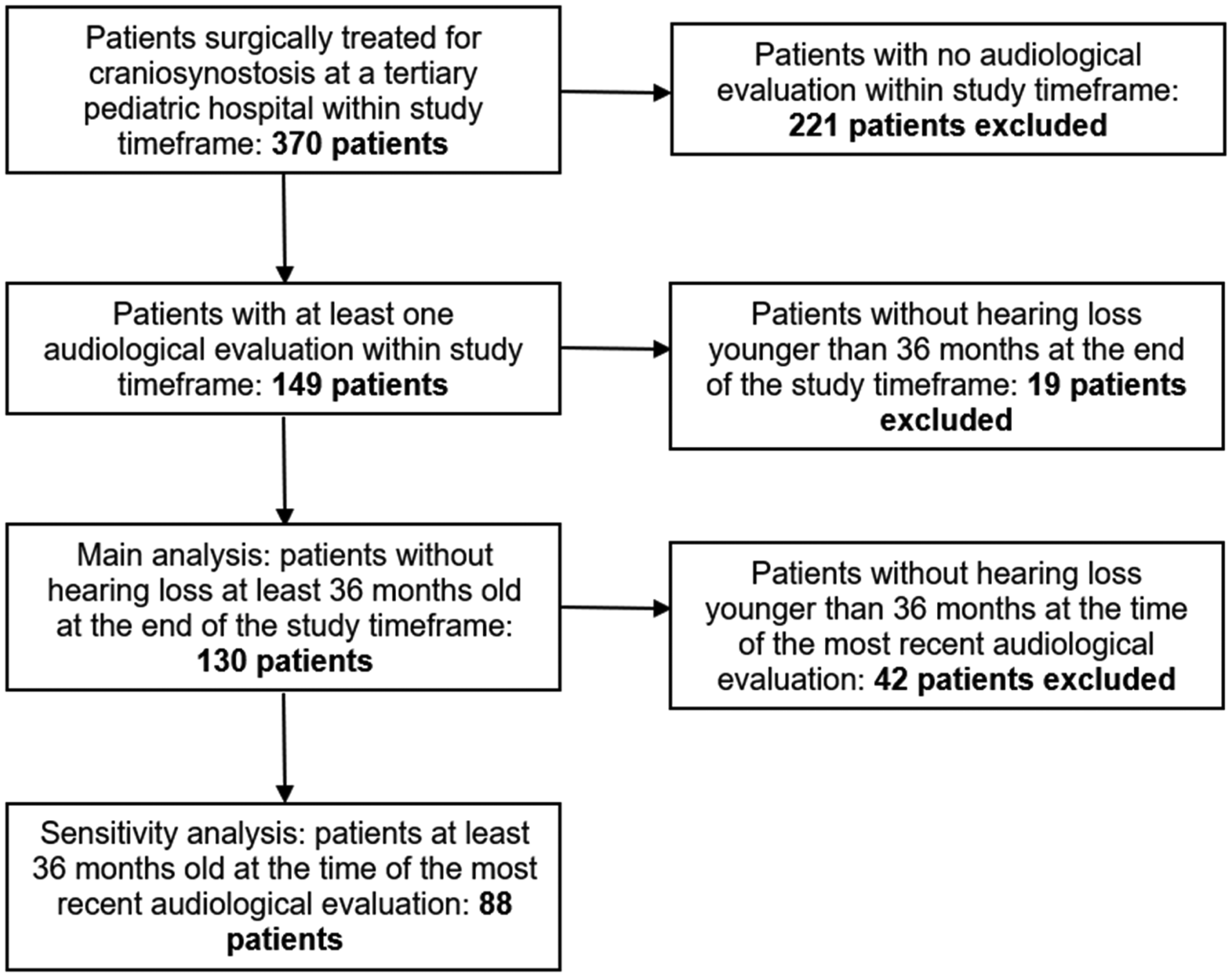
Inclusion criteria for the study.
Coder Reliability
Documentation from plastic surgery, neurosurgery, genetics, otolaryngology, and audiological specialties were reviewed by three trained coders, including one primary coder who trained two secondary coders on extracting relevant data from the electronic medical record for each variable. Incorrect data points were reviewed, and the primary coder provided feedback to ensure consistency. Consistency among all raters was evaluated based on the first ten patients for all variables in the study using Cohen's Kappa (k > .89).
Variables of Interest
Demographic variables included gender, age at the end of the study, race, ethnicity, primary language, and use of private insurance. Family history of craniosynostosis, speech language disorder or delay, and craniofacial or genetic syndrome were also recorded. Key variables included anatomical type of craniosynostosis, syndromic status, and medical complexity. Syndromic status was initially determined by clinical evaluation with all patients then referred to genetic testing. Of the 23 patients with genetic syndromes related to craniosynostosis, 19 (82.6%) were confirmed by genetic testing, and the other four patients were clinically diagnosed with Apert syndrome. Patients were considered non-syndromic if genetic testing confirmed a syndrome that was not related to craniosynostosis (eg, 22q deletion syndrome, 6q deletion syndrome, VACTERL syndrome, CHARGE syndrome, etc.) or if there were no clinical evidence of a syndrome. Patients were included in the medically complex category if they had a diagnosed syndrome of if they had any of the following medical comorbidities which likely increased the risk for hearing loss: presence of a cleft palate, tracheostomy, major cardiac defect requiring cardiac surgery, significant vision impairment, global developmental delay, ventriculoperitoneal shunt, Chiari 1 malformation, or epilepsy. Therefore, the primary three variables that are suspected of affecting hearing loss are the type of craniosynostosis, presence of a genetic syndrome, and medical complexity.
Coding of Hearing Data
Audiology evaluation notes were reviewed, and the following data points were extracted: results of newborn hearing screening; number of audiograms completed; whether testing was completed behaviorally in a Soundfield, with headphones, or as an auditory brainstem response (ABR); and the presence and laterality of hearing loss. Hearing loss was categorized into two categories: CHL and permanent hearing loss. CHL was defined as hearing loss due to a temporary elevation of thresholds due to otitis media that resolved with medical treatment. Permanent hearing loss was defined as any SNHL or MHL. These two categories were chosen given the different long-term hearing prognoses based on these diagnoses. A single visit with an audiologist and audiogram was sufficient to determine hearing status, as the testing battery pediatric audiologists use provide a differential diagnosis and robust cross-checking for hearing loss. To qualify for CHL, the patient had to have evidence of it or middle ear fluid on tympanometry. While many of these children had further evidence of normal hearing after medical management (eg, placement of tympanostomy tubes), this was not routinely done at our institution given the high volume of audiological appointments. To qualify for permanent hearing loss, the patient had to have evidence of elevated bone conduction thresholds consistent with a sensorineural component. Additionally, if the child used amplification, the type of amplification used (traditional hearing aid, bone conduction device, and cochlear implant) was extracted from the electronic medical record. Otolaryngology notes were also reviewed to determine history of tympanostomy tube placement and number of tympanostomy tubes placed.
Statistical Analysis
Median, interquartile range (IQR), frequency, and percent were used to summarize demographics, family history, patient medical history, types of genetic syndromes, and hearing status. Outcomes of interest included CHL (vs normal hearing) and permanent hearing loss (vs normal hearing). Wilcoxon rank-sum tests were used to compare groups for continuous variables, and Chi squared or Fisher's exact tests were used to evaluate frequency distributions. Logistic regression models were fit for each outcome. For categorical predictors with more than two levels, the likelihood ratio test was used to compare the full model to the null model. A single multivariable model predicting each outcome was evaluated for three factors of interest: syndromic status, anatomical type of craniosynostosis, and medical complexity. Genetic syndromes were included given their reported relationship with hearing loss in the literature. Non-syndromic craniosynostosis was separated by its anatomical types to understand if the relationship of which cranial suture prematurely fused affected hearing loss. Medical complexity was included to control for other common causes of hearing loss (eg, cleft palate, middle ear effusion, etc.), other factors due to syndromes that are unrelated to craniosynostosis, or other significant medical comorbidities. Age in months was used as a covariate when this was significant in the univariable analysis. Due to small sample size and sparse data, for univariable models only, Firth penalized maximum likelihood method (from the logistf package in R) was used to improve accuracy and stability of odds ratio (OR) estimates and ensure model convergence when data separation issues occurred. All models were repeated in the sensitivity analysis to determine if they were robust to stricter inclusion criteria. Model results were reported using unadjusted ORs and adjusted ORs, 95% confidence intervals (CIs), and P-values. P-Values less than .05 were considered significant. All analyses were conducted using R version 4.3.0.
Results
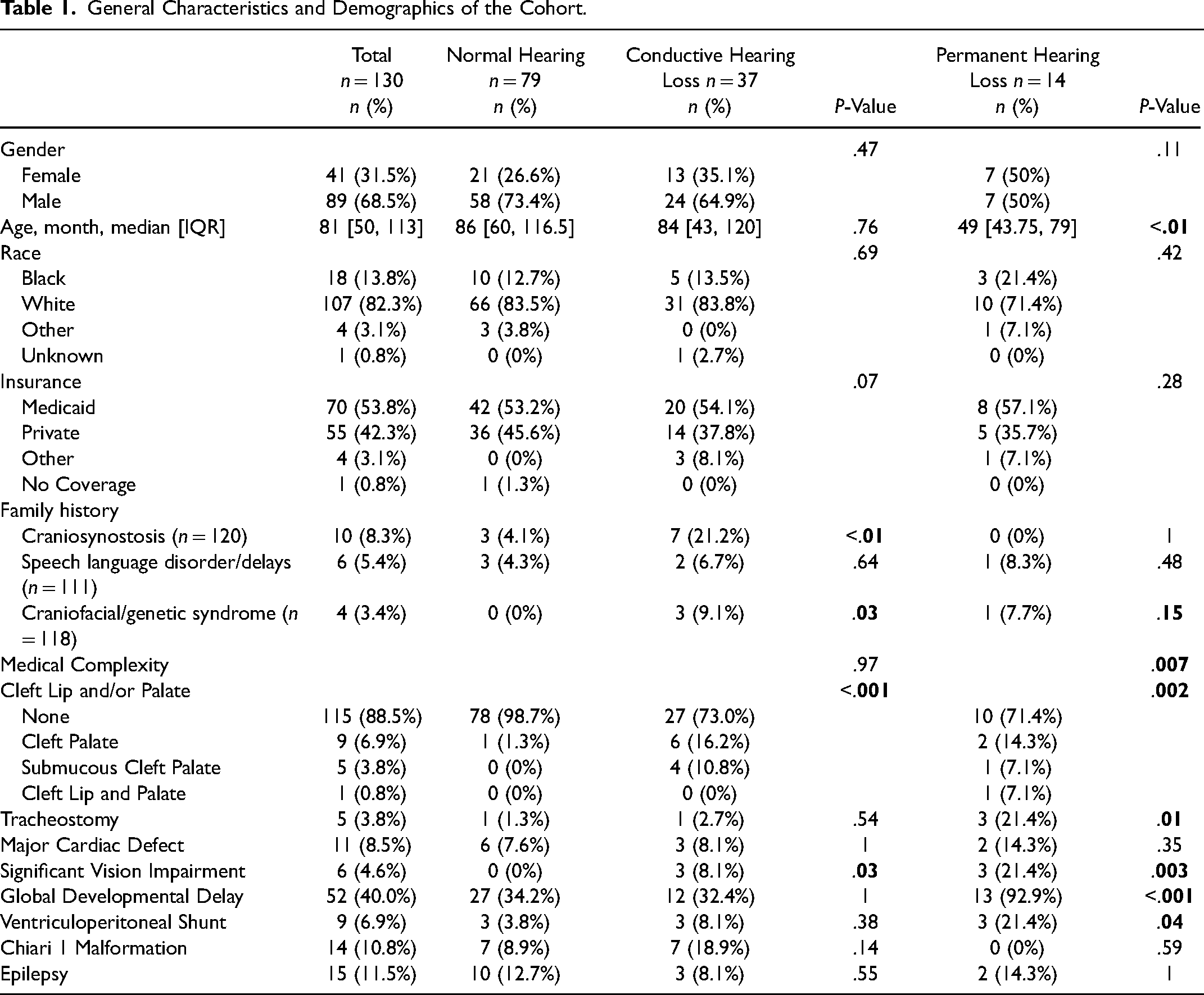
Of the 370 patients with craniosynostosis, the final sample included 130 patients: 79 with normal hearing, 37 with CHL, and 14 with permanent hearing loss. The sensitivity analysis sample included 88 patients: 37 with normal hearing, 37 with CHL, and 14 with permanent hearing loss. Demographics and the components of medical complexity based on patients’ other medical comorbidities are shown in Table 1. Unsurprisingly, patients with cleft palate and those with significant medical comorbidities (eg, significant vision impairment, tracheostomy, global developmental delay, or ventriculoperitoneal shunt) presented with hearing loss more frequently. Table 2 presents the anatomical types of craniosynostosis and genetic syndromes associated with craniosynostosis across children with normal hearing and conductive and permanent hearing loss. Forty-five patients presented with sagittal (34.6%), 28 metopic (21.5%), 25 multisuture (19.2%), 16 bicoronal (12.3%), 12 unicoronal (9.2%), and four lambdoid (3.1%) craniosynostosis. Twenty-three patients presented with syndromic craniosynostosis (17.7%) and 107 with non-syndromic craniosynostosis (82.3%). Syndromes included 8 patients with Apert (34.7%), 5 with Crouzon (21.7%), 4 with Muenke (17.4%), 2 with Antley-Bixler (8.6%), 1 with Saethre-Chotzen (4.3%), 1 with Jacobsen (4.3%), 1 with Carpenter (4.3%), and 1 with Pfeiffer (4.3%) syndrome. Of the 23 patients with syndromic craniosynostosis, 5 patients (21.7%) had permanent hearing loss, 16 patients (69.6%) had CHL, and 2 patients (8.7%) had normal hearing. Table 2 also presents additional audiological variables included in the study. The median number of audiograms was 1 [IQR:1,2] for those with normal hearing, 3 [IQR:2,5] for those with CHL, and 5.5 [IQR:4,7.8] for those with permanent hearing loss. Patients who failed their newborn hearing screen were also more likely to present with conductive (76.7% pass) or permanent (38.5% pass) hearing loss than those with normal hearing (93.4% pass). Additionally, 100% (14/14) of patients with permanent hearing loss received hearing aids (eg, bone conduction devices or cochlear implants). Lastly, the placement of tympanostomy tubes corresponded with the rate of CHL (for recurrent acute otitis media [AOM], P = .007).
General Characteristics and Demographics of the Cohort.
Hearing Loss Across Types of Craniosynostosis and Genetic Syndromes.
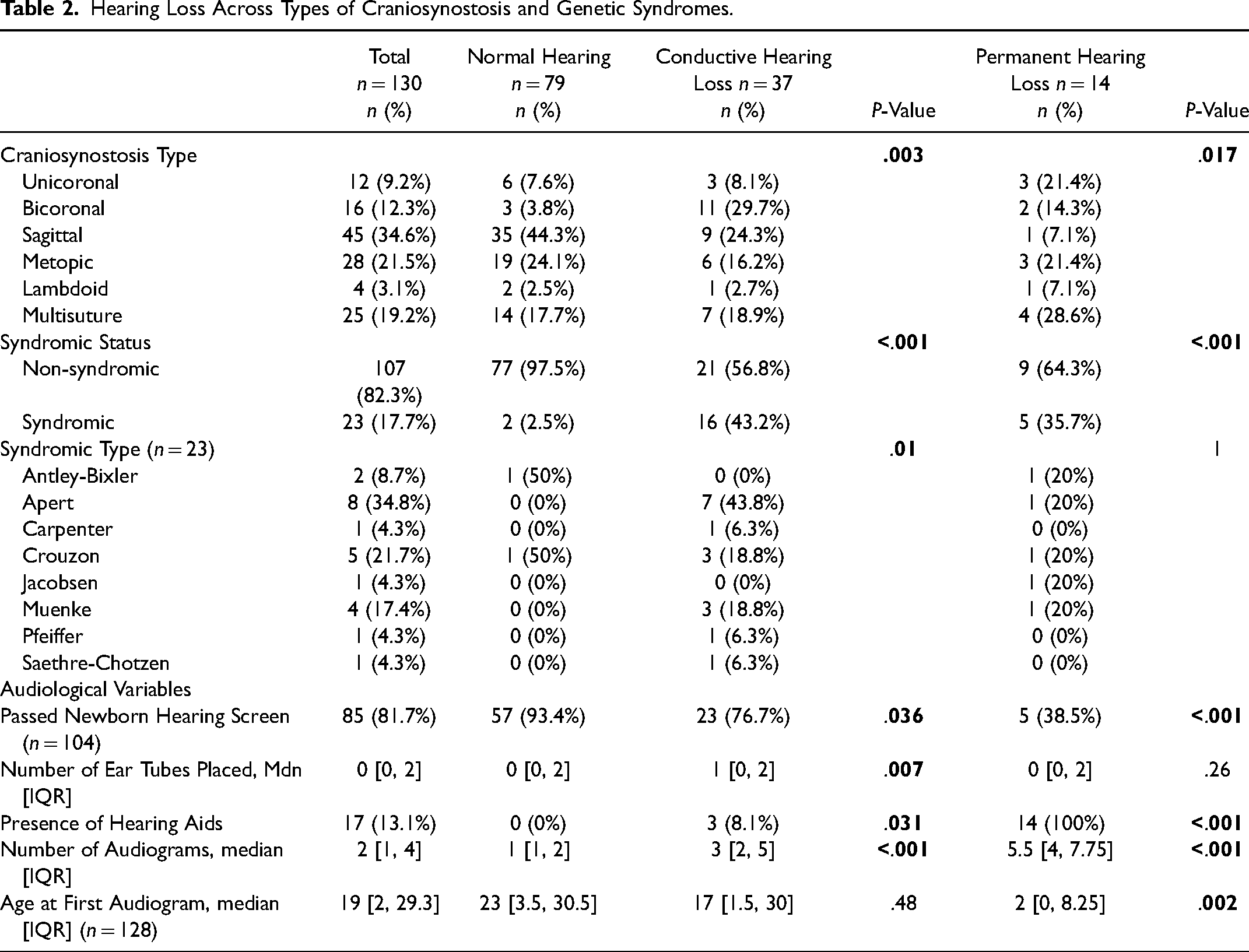
Table 3 shows the univariable analysis for hearing loss in the main sample (n = 130). The only demographic variable that was a significant predictor for permanent hearing loss was age (0.997 OR, P = .01). Family history of craniosynostosis (5.78 OR, P < .01), family history of a craniofacial syndrome (16.64 OR, P = .02), and patient history of cleft palate (19.98 OR, P < .001) increased the odds for CHL. Patients with medically complex histories did not have higher odds for CHL (P = .81) but had higher odds for permanent hearing loss (10.88 OR, P = .03). Bicoronal and syndromic craniosynostosis both had higher odds for conductive (14.26 OR, P < .001 and 29.33 OR, P < .001, respectively) and permanent hearing loss (23.33 OR, P = .02 and 21.39 OR, P = .001, respectively). Unicoronal (17.50 OR, P = .02) and multisuture (10.00 OR, P < .05) craniosynostosis had higher odds for permanent hearing loss. Craniosynostosis type overall was a significant predictor of conductive (P < .01) and a marginal predictor of permanent (P = .05) hearing loss.
Main Analysis of Univariable Logistic Regression for Demographics, Family History, Patient History, Type of Craniosynostosis, and Genetic Syndrome With Hearing Loss Outcomes.
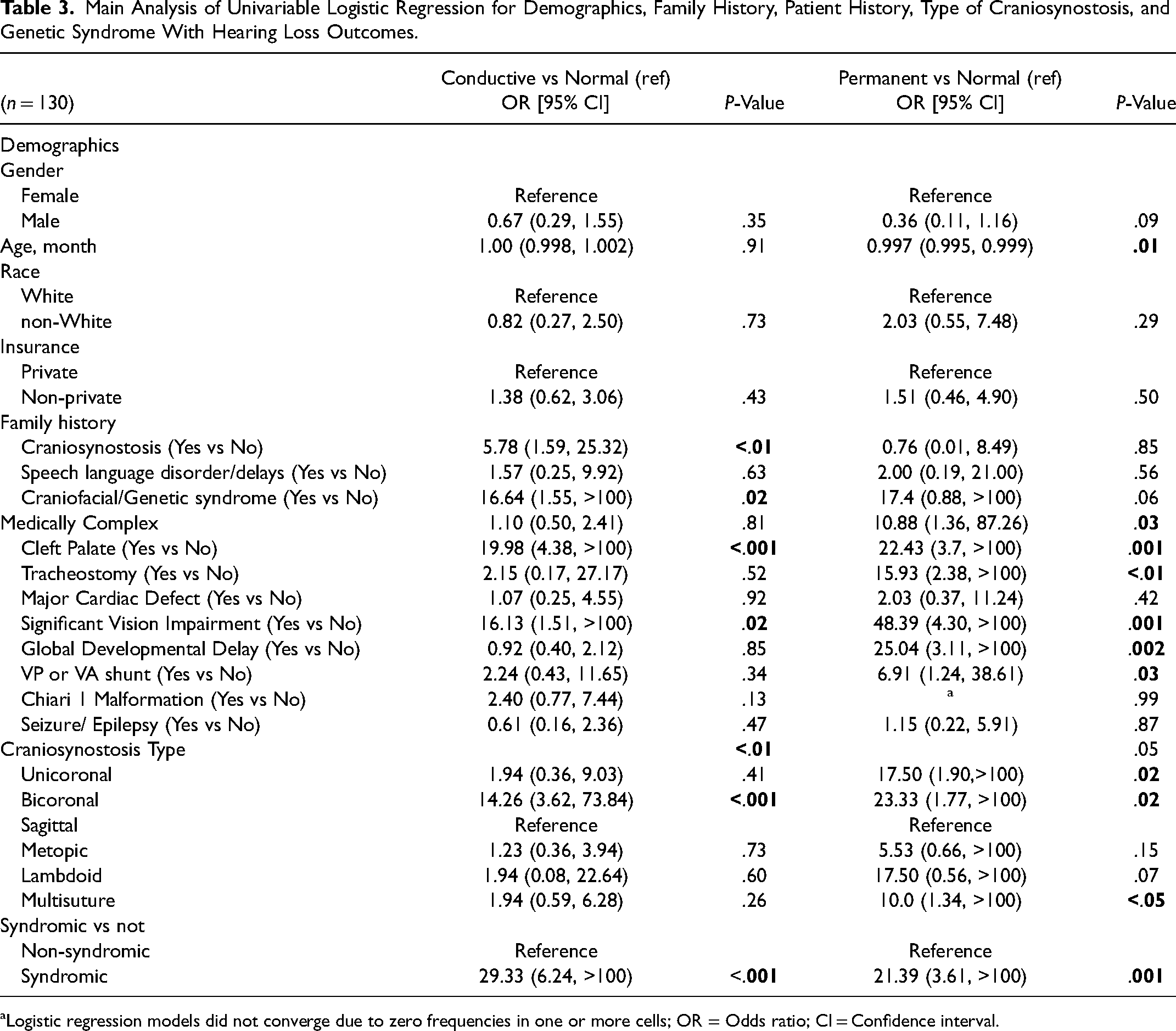
Logistic regression models did not converge due to zero frequencies in one or more cells; OR = Odds ratio; CI = Confidence interval.
Table 4 shows the univariable analysis for hearing loss in the sensitivity analysis (n = 88). Here, age was a significant predictor for both conductive (0.98 OR, P < .05) and permanent (0.994 OR, P = .001) hearing loss. Family history of craniosynostosis was no longer a significant predictor of CHL, though patients with significant other medical comorbidities presented with higher odds for permanent hearing loss (8.86 OR, P = .045). Bicoronal and syndromic craniosynostosis were both still significant for higher odds for conductive (5.30 OR, P = .03 and 27.43 OR, P = .002, respectively) hearing loss. Unicoronal (19.50 OR, P = .03) and syndromic (20.00 OR, P = .01) craniosynostosis had higher odds for permanent hearing loss, though bicoronal and multisuture craniosynostosis were no longer significant for permanent hearing loss. Craniosynostosis type overall was no longer a significant predictor of conductive (P = .25) and permanent (P = .23) hearing loss.
Sensitivity Analysis of Univariable Logistic Regression for Demographics, Family History, Patient History, Type of Craniosynostosis, and Genetic Syndrome with Hearing Loss Outcomes.
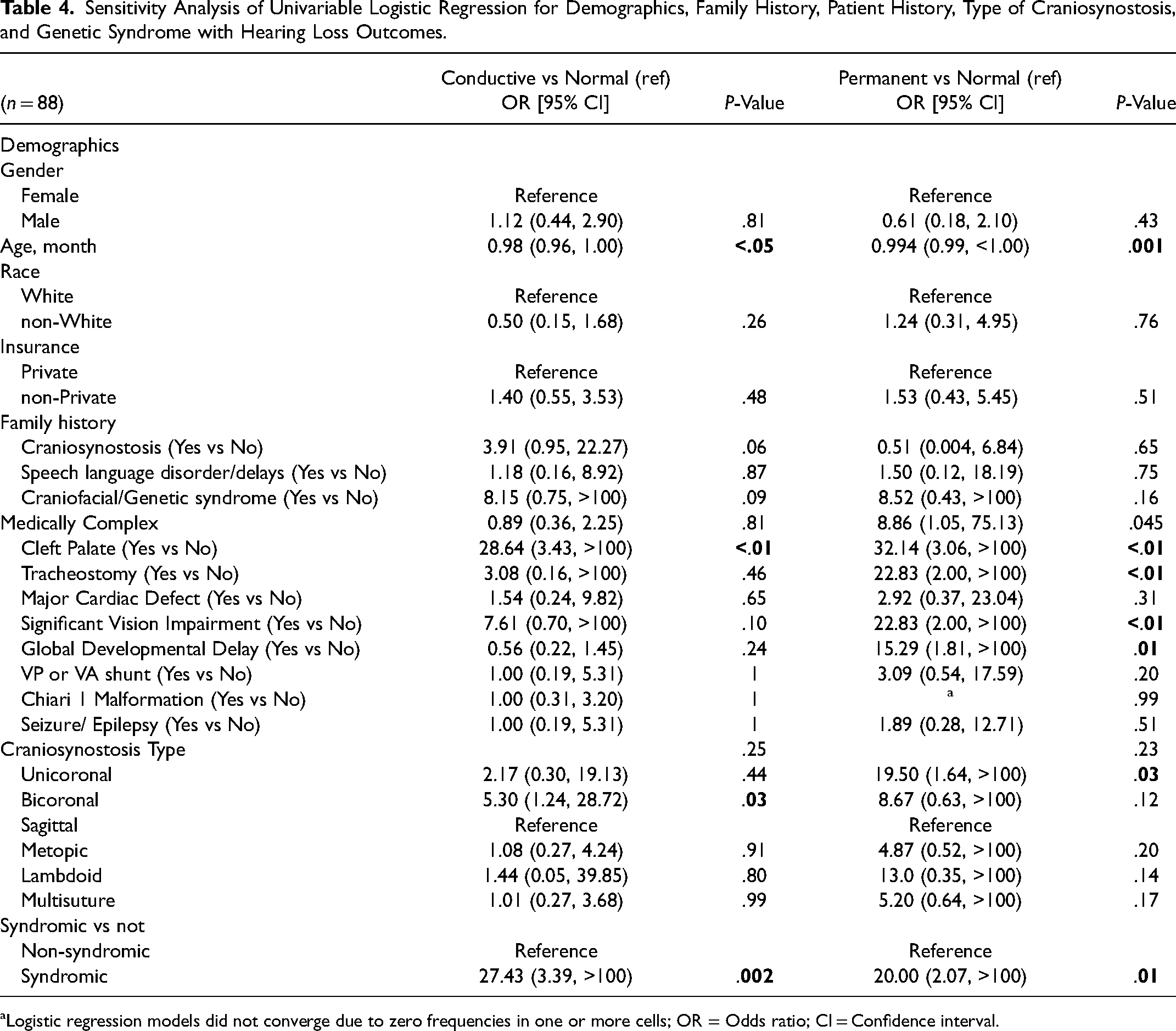
Logistic regression models did not converge due to zero frequencies in one or more cells; OR = Odds ratio; CI = Confidence interval.
In the multivariable analysis shown in Table 5, the top panel shows the patients in the main analysis (n = 130). Patients with syndromic craniosynostosis (30.13 OR, P = .03), unicoronal craniosynostosis (16.19 OR, P < .05), and those with significant medical comorbidities (22.06 OR, P = .02) had higher odds for permanent hearing loss, whereas only syndromic craniosynostosis (45.94 OR, P = .001) had higher odds for CHL. The bottom panel of Table 5 shows the multivariable analysis for the sensitivity analysis (n = 88). Statistical significance held with the more restrictive normal hearing inclusion criteria for syndromic craniosynostosis (34.51 OR, P < .05) and those with a medically complex history (23.36 OR, P < .05) for permanent hearing loss and only syndromic craniosynostosis (49.80 OR, P = .002) for CHL. However, unicoronal craniosynostosis was no longer significant for permanent hearing loss (P = .09) in the sensitivity analysis.
Multivariable Logistic Regression: Main and Sensitivity Analysis.
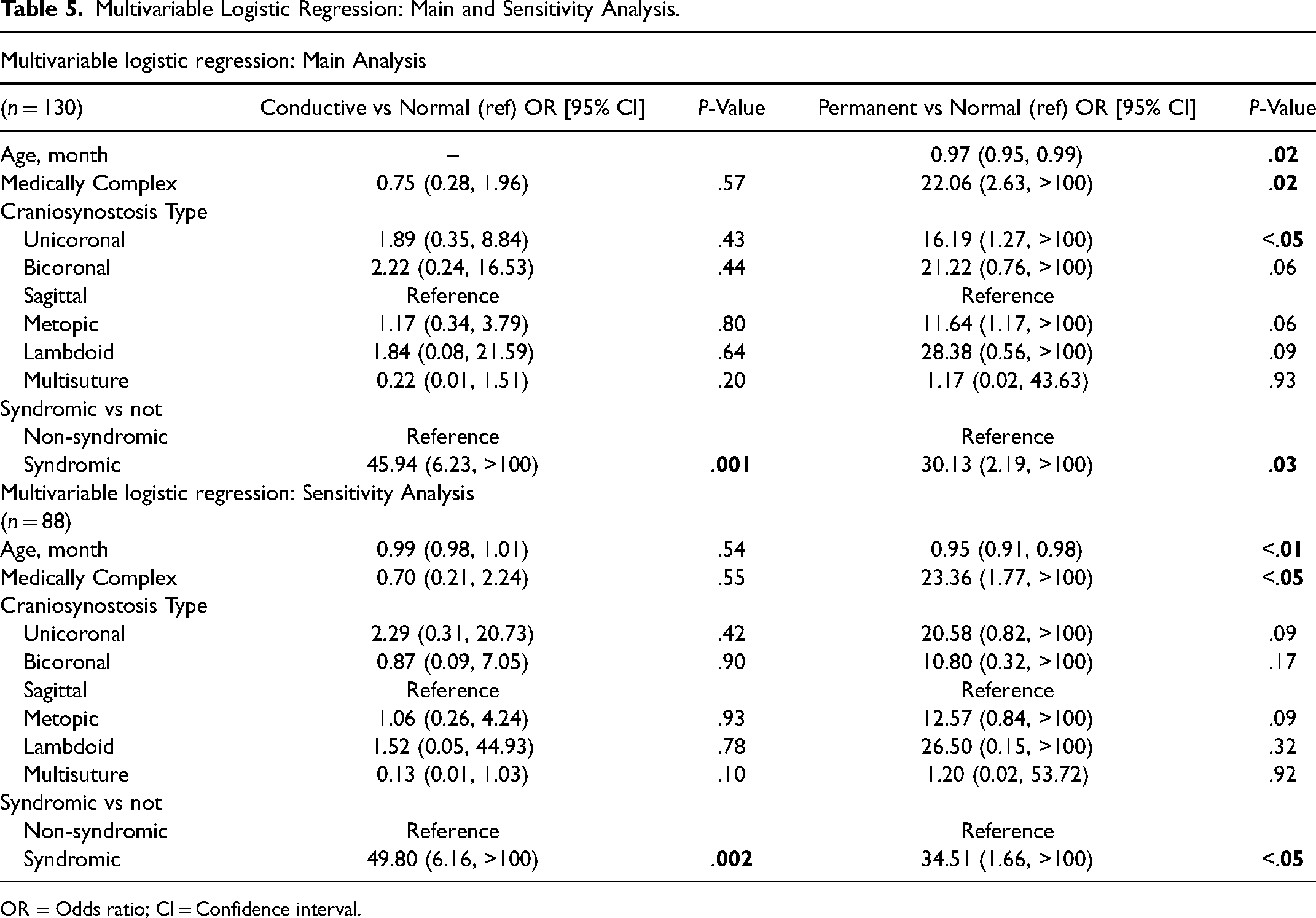
OR = Odds ratio; CI = Confidence interval.
Discussion
Logistic regression models evaluated the effects of demographics, family history, patient medical history, and characteristics of craniosynostosis on hearing loss outcomes. A multivariable model focused on three key factors associated with conductive or permanent hearing loss: medical complexity, anatomical type of craniosynostosis, and syndromic status. In this study, syndromic craniosynostosis had the largest and most consistent effect on permanent and CHL across all models with 91.3% of patients with syndromic craniosynostosis presenting with hearing loss. This is consistent with several studies in which multiple craniosynostosis syndromes have been associated with hearing loss.15–18,20,21 In most of these syndromes, hearing loss cases are reported as conductive rather than sensorineural (or permanent). Most of our 23 patients with syndromic craniosynostosis had conductive rather than permanent hearing loss. Although Muenke syndrome has classically been described as having SNHL, some studies have found more conductive rather than permanent hearing loss.20,21 Our results with Apert syndrome showed 87.5% of patients having CHL. The literature describes Apert syndrome as commonly presenting with CHL either due to middle ear effusion or congenital stapes fixation. 25 While the cases of middle ear effusion can explain the CHL, congenital stapes fixation often presents with permanent hearing loss as surgery is not consistently completed due to variable outcomes, though our one patient with permanent hearing loss and Apert syndrome did not have congenital stapes fixation. Our results also showed primarily CHL for Carpenter, Crouzon, Pfeiffer, and Saethre-Chotzen syndromes versus permanent hearing loss for Antley-Bixler and Jacobsen syndromes. Carpenter syndrome has been hypothesized to have increased intracranial pressure from cranial anomalies which may predispose patients to frequent otitis media and hearing loss. 26 In Antley-Bixler and Jacobsen syndromes, hearing deficits are not well described but have been reported.27,28 Lastly, for Crouzon, Pfeiffer, and Saethre-Chotzen syndromes, hearing loss has been described as a result of changes in skull structure and recurrent otitis media leading to CHL.15,29,30 Overall, in our study, patients with syndromic craniosynostosis had significant odds for conductive and permanent hearing loss.
In our study, 28% of patients with non-syndromic craniosynostosis presented with hearing loss. There is limited literature on rates of hearing loss among those with non-syndromic craniosynostosis. Grewal et al found that, 9% of 133 patients with non-syndromic craniosynostosis had hearing loss. 22 They also noted that 36% had OME on either history, physical examination, tympanometry, or imaging. 22 Prager et al found that 26% of 57 non-syndromic craniosynostosis patients had hearing loss with 80% of these cases being CHL, attributing most of this to middle ear effusion. 24 While it is challenging to determine the rate of hearing loss in general pediatric populations, Paul et al notes that roughly 50% of all children will have at least one ear infection by 24 months of age, 31 and Meherali et al found that AOM is most common between 6 and 24 months of age.32,33 Vergison et al states that more than 80% of children will develop AOM at least once before 3 years of age. 34 While AOM was likely the most common cause of CHL in our study, it is difficult to convert the prevalences of AOM given by Paul, Meherali, and Vergison et al into CHL detected at a given audiological appointment (28.5% in our study). It is unclear what mechanism might explain hearing loss in non-syndromic craniosynostosis outside of the more common middle ear effusion. While syndromic craniosynostosis cases have proposed mechanisms of changes in skull structure predisposing to increased rates of hearing loss, further studies are needed to examine whether there are similar mechanisms at play for non-syndromic craniosynostosis. Only through a prospective study in which all patients with craniosynostosis regardless of syndrome had serial audiological evaluations over time throughout early childhood could this be determined. Currently, recommendations for audiological testing is on an as needed basis rather than required for all patients with craniosynostosis.8,35
As for the anatomic type of craniosynostosis, it was likely that the univariable models captured the effect of syndromic craniosynostosis for hearing loss since unicoronal, bicoronal, and multisuture craniosynostoses are more commonly associated with syndromes than metopic, lambdoid, or sagittal craniosynostoses. However, unicoronal craniosynostosis had higher odds for permanent hearing loss in the multivariable model, which controlled for syndromes. Likewise, certain anatomical types of craniosynostosis approached significance in the multivariable model for permanent hearing loss: bicoronal (P = .06), metopic (P = .06), and lambdoid (P = .09) craniosynostoses. This suggests that an alternative mechanism for hearing loss is at play for these types of craniosynostosis, and further studies describing this are warranted.
Medical complexity was included in the multivariable model to control for other causes of hearing loss aside from type of craniosynostosis and genetic syndrome. While not comprehensive, it included other possible causes of hearing loss like cleft palate as well as many congenital anomalies that are commonly seen in syndromes. Medical complexity remained significant in the multivariable model for permanent hearing loss (P < .05) but not for CHL (P = .55). This increases the internal validity of medical complexity since congenital causes of hearing loss should present with permanent hearing loss rather than CHL, which would more commonly be associated with middle ear effusion.
To ensure that our models were valid, supplemental analyses of audiological variables of our cohort were completed (Table 2). Patients with permanent and CHL had a greater median number of audiograms than patients with normal hearing, suggesting that patients with more severe hearing loss received additional follow-up compared with their normal hearing counterparts. Additionally, it was likely that a majority of the patients with permanent hearing loss in our study had it since birth as patients who failed their newborn hearing screen were more likely to present with permanent hearing loss. These patients with permanent hearing loss were all successfully referred and managed, as 14/14 received hearing aids (eg, bone conduction devices or cochlear implants). Lastly, while the placement of tympanostomy tubes corresponded with the rate of CHL (ie, for recurrent AOM), routine audiograms after the placement of tympanostomy tubes—or after any diagnosis of CHL—were not conducted at our institution since volumes are too high to conduct this practically, which present as a limitation in our study.
As for additional limitations, there was a reduction in sample size for the main analysis, sensitivity analysis, and among the number of syndromic craniosynostosis cases. The main reason for the sensitivity analysis was the worry of loss to follow-up. Our objective was to ensure that patients had sufficient time to develop hearing loss, hence the restrictions for age at the end of the study (main analysis) and age at the most recent audiological evaluation (sensitivity analysis). The main and sensitivity analyses protect against the inherent bias that exists when patients are normally tested for hearing loss. This is because not every patient is screened for hearing loss after the newborn hearing screen and patients are more likely to be screened if they present with symptoms of hearing loss. The sensitivity analysis increased our confidence in the results that remained significant and created a more uniform population of patients with normal hearing. While having two separate analyses is a limitation and can introduce bias, it also adds robustness for effects that remained statistically significant and balances validity with more rigorous inclusion criteria and statistical power with greater sample size across the two models. Given that this study was a retrospective cohort study, another key limitation is selection bias resulting from patients with concerns for hearing loss being more likely to receive an audiological evaluation. In this way, the sample may not be representative of the general population of children with craniosynostosis. It is likely that children with trisomy 21 (who reportedly have at least a three times higher incidence of chronic otologic disease compared with normally developing children), 36 cleft lip and/or palate (studies report at least 90% of patients with cleft palate with middle ear disease and that the incidence of hearing loss in patients with cleft lip and palate ranges up to 83%),37–39 middle ear effusion, speech language delays, or syndromes known to increase risk for hearing loss were more frequently tested, which does not include many of the children with craniosynostosis without referral for audiological testing. Therefore, this biases our cohorts with an underrepresentation of patients with normal hearing. While the inclusion of medical complexity in the multivariable model was intended to limit this bias and other causes of hearing loss in our study, it is possible that the percentages of hearing loss estimates in this study are overestimated. Additionally, without a control group in this retrospective cohort study that underwent the same audiologic protocol, this study cannot compare prevalence and profiles of hearing loss in children with craniosynostosis to children without this condition. Finally, it is important to note that ORs from logistic regression models should be interpreted with caution. Our interpretation focused on the direction of effects (increased or decreased odds for the outcome) rather than the magnitude of effects since ORs are exaggerated due to sparse data bias (small sample size). Ultimately, future studies would benefit from combining data from multiple centers with audiological evaluations, which would also be more representative of the subset of patients who have comorbidities of hearing loss and specific genetic syndromes given the increase in sample size and greater diversity of patient populations.
From this study, we conclude that all children with syndromic craniosynostosis and those with medically complex histories should be referred to audiology for evaluation of hearing loss. Furthermore, it is possible that children with non-syndromic craniosynostosis or those without significant medical comorbidities are at higher risk of hearing loss, warranting audiological screening as well. Nevertheless, further prospective studies are recommended to determine the prevalence of hearing loss in patients diagnosed with craniosynostosis.
Conclusions
In a retrospective cohort study at a tertiary pediatric hospital, 130 children diagnosed with craniosynostosis with adequate audiological follow-up were studied, of which 28.5% (37/130) presented with CHL and 10.8% (14/130) presented with permanent hearing loss. Multivariable analysis of syndromic status, anatomical type of craniosynostosis, and medical complexity revealed that patients with syndromic craniosynostosis, those with significant medical comorbidities, and those with unicoronal craniosynostosis have significantly higher odds for permanent hearing loss whereas patients with syndromic craniosynostosis had higher odds for CHL. The consistency of these results across the multivariable and sensitivity analysis models suggest that these findings are robust. Additionally, 28% of patients with non-syndromic craniosynostosis had evidence of hearing loss. Larger multi-center prospective studies are necessary to characterize rates and type of hearing loss to avoid the potential risk of selection bias. The preliminary data from this study suggests that children with syndromic craniosynostosis warrant audiologic testing and monitoring. Further study of children with non-syndromic craniosynostosis is needed to better identify other medical comorbidities and predictors associated with risk for hearing loss and if similar audiologic surveillance is indicated.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.