
Abstract
Objective
This study aimed to identify specific causes, clinical features, and contributing factors associated with early childhood mortality in a vulnerable subset of patients with cleft lip and/or palate (CL/P) who did not survive to undergo surgical repair.
Design
This was a retrospective chart review study.
Setting
The study was conducted at a single tertiary medical center.
Patients/Participants
We identified 26 liveborn CL/P patients who died without cleft repair from 1970 to 2024. Their data was compared to a control group of 2070 surgically repaired CL/P patients from the same institution.
Main Outcome Measures
Our primary goal was to characterize the clinical profile of patients experiencing prerepair mortality. Secondary outcomes included identifying causes of death, maternal and prenatal risk factors, and comparing these findings with the surgically repaired patients.
Results
All 26 unrepaired patients (100%) had genetic syndromes or isolated severe congenital anomalies, a significant difference compared to 24% in the surgically repaired group (P < .0001). Trisomy 13 (30.8%), holoprosencephaly (23.1%), congenital cardiac defects (11.5%), and trisomy 18 (11.5%) were the leading causes of death. The median age at death for unrepaired patients was 25 days (interquartile range 4-125 days), significantly lower than the 61.9 years in the control group (P < .0001). Multivariable logistic regression showed that genetic syndromes or severe congenital anomalies were strongly associated with early mortality (P < .0001). Notably, all unrepaired patients who were born in the United States after 2005 had accurate prenatal diagnoses, which allowed clinical planning before the patients were born.
Conclusions
Prerepair mortality in CL/P is significantly driven by underlying genetic syndromes or congenital anomalies. For surgeons, this highlights the importance of genetic testing and prenatal care to guide appropriate surgical management. Considering a wait in surgical intervention until patients are approximately 4 months old may allow for better survival assessment and informed decisions for these high-risk individuals.
Introduction
Despite advancements in modern medicine and surgical techniques, cleft lip and/or cleft palate (CL/P) can still be associated with early childhood mortality in a small subset of patients.1–4 While surgical repair remains the definitive treatment for CL/P cases, enabling them to achieve normal life expectancy, a vulnerable group of infants does not survive long enough to undergo these corrective procedures.4–7 We did not find dedicated and detailed publications specifically focusing on this population with mortality before surgical repair. Some related information can be found in studies of the overall mortality rate in CL/P patients, mostly from European countries with more comprehensive registries and population-based birth data. 8 These studies suggest that prerepair mortality is rarely the direct result from cleft itself but is instead frequently linked to severe co-occurring congenital defects or genetic syndromes.1–3,5,9–11 For instance, trisomy 13 (Patau syndrome) is identified as a leading cause, due to its systemic anomalies including cardiac and central nervous system defects.1,10,12 Other contributing factors, including feeding difficulties and low birth weight, further complicate the readiness for surgery.3,13,14 There are also ethnical concerns regarding whether these children should undergo surgical repair, balancing the risks of medical intervention with potential benefits for their quality of life.4,5,15
This study aims to investigate the specific causes, clinical features, and contributing factors associated with prerepair deaths in CL/P. We seek to provide clinicians with specific information to identify high-risk individuals earlier, customize care, and offer accurate counseling to families. Furthermore, this research can inform decision-making regarding choice for surgical intervention for patients with complex birth defects, ensuring that medical care aligns with the best interests of each child and their family.
Patients and Methods
Study Design
This retrospective study utilized the comprehensive electronic health records at our institution to identify patients from January 1970 to the end of 2024 with a confirmed diagnosis of CL/P. Initial diagnoses were established by screening relevant International Classification of Diseases, Ninth and Tenth Revision (ICD-9 and ICD-10) codes in the database. Within this identified CL/P cohort, we then specifically searched for individuals who passed away in their childhood without any record of surgical repair for their CL/P (confirmed by procedural codes and thorough review of clinical notes). The study protocol was approved by the Institutional Review Board at our institution. Informed consent has been waived.
Case Ascertainment and Study Variables
For the cases of prerepair mortality, we included patients who had sufficient medical records for review for at least their first year of life or until death, whichever occurred first. We excluded those earlier patients who did not have adequate documentation or if the handwritten medical records were uninterpretable. We also excluded patients whose deaths were documented as stillborn or intrapartum death (Figure 1). The control group for comparison was defined as liveborn patients with a confirmed diagnosis of CL/P who underwent cleft repair surgery at our institution.

Patient Screening Flowchart.
Key variables collected included patient demographics (eg, sex, race/ethnicity), the specific cleft subtype, gestational age at birth (birth weeks), birth weight, prenatal information, age at death, cause of death, and co-occurring birth defects. Relevant maternal pregnancy and prenatal care history were also collected.
Outcomes
The primary outcome of this study was to characterize the clinical features of patients with CL/P who experience prerepair mortality. Secondary outcomes included the identification of contributing causes of death, and the assessment of relevant maternal characteristics and prenatal history. Another key aspect of our investigation was to compare the clinical features and associated congenital anomalies of CL/P patients who experienced prerepair mortality with those who underwent cleft repair surgery.
Statistical Analysis
Descriptive statistics were used to summarize demographic and clinical characteristics. Continuous variables were presented as means and standard deviations (mean ± SD) if normally distributed, or as medians and interquartile ranges (IQRs) for skewed distributions. Categorical variables were reported as frequencies and percentages.
For comparisons between the prerepair mortality group and the control group, differences in continuous variables were examined using Mann–Whitney U test. Differences in categorical variables were evaluated using chi-squared test. Multivariable logistic regression was applied for identifying predictors, providing odds ratios (ORs) and 95% confidence intervals (CIs). P values <.05 were considered statistically significant.
All statistics were conducted in GraphPad Prism software (GraphPad v10.0, CA). This study is consistent with the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines.
Results
Patient Characteristics
A total of 26 liveborn individuals were included, born between 1976 and 2024. The cohort consisted of 15 boys and 11 girls. The majority of patients were White, comprising 88.5% (n = 23) of the cohort, with 3 individuals of East Asian descent. The median age at death for these children was 25 days (IQR 4-125 days), with only 3 patients surviving beyond 1 year of age. The median birth week for this cohort was 36 + 3 weeks (IQR 32 + 2 to 38 + 5 weeks), with 58% (n = 15) of patients born preterm. The average birth weight was 2198.05 ± 961.53 g. Furthermore, 65.4% (n = 17) of patients had birth weights below the 3rd percentile. The average Apgar scores were 4 ± 3 at 1 min and 7 ± 2 at 5 min, while a normal Apgar score is considered to be 7 or higher at both timepoints. All 26 patients (100%) were diagnosed with either a genetic syndrome (eg, trisomy 13, CHARGE syndrome) or isolated severe congenital anomalies (eg, tetralogy of Fallot, renal agenesis, transposition of great arteries). In this study, we defined congenital anomalies as isolated major structural or functional abnormalities that could not be attributed to a specific syndromic diagnosis. Notably, 73% (n = 19) of the patients had both cleft lip and palate.
Of the deaths in this cohort, 25 (96.2%) were documented due to complications from a genetic syndrome or congenital abnormality, rather than directly from CL/P. One death which occurred back in 1983, at the age of 35 days, was caused by aspiration of fluids into the lungs from swallowing difficulties. This event could have been related to feeding difficulties from the cleft palate, but given the neurological and cardiac comorbidities, it can also be multifactorial. Among the primary causes of death, trisomy 13 ranked as the number 1 cause, followed by holoprosencephaly and severe cardiac defects. Some patients had both a trisomy diagnosis and cardiac or central nervous system defects. These individuals were categorized under the trisomy group. Patient characteristics are detailed in Table 1.
Patient Demographics and Clinical Characteristics.
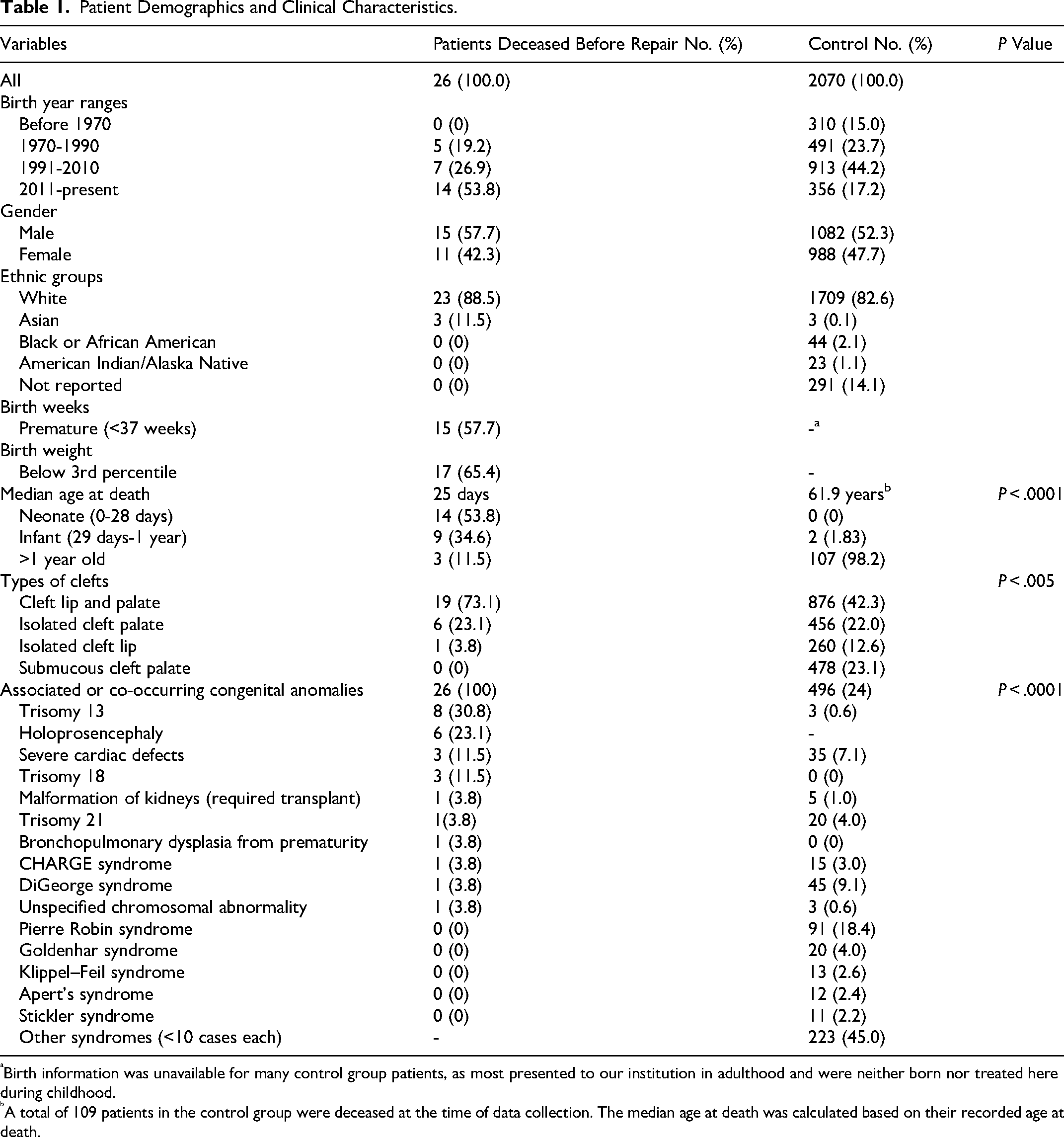
Birth information was unavailable for many control group patients, as most presented to our institution in adulthood and were neither born nor treated here during childhood.
A total of 109 patients in the control group were deceased at the time of data collection. The median age at death was calculated based on their recorded age at death.
While most patients did not survive until the recommended age for surgical repair, 7 patients lived beyond 3 months of age (ranging from 125 to 1166 days). Of these, only 1 patient underwent cleft lip repair but not cleft palate repair while the remaining 6 did not undergo any cleft repair at all. The reasons for not having surgery, even when reaching the indicated age from ENT or plastic surgery consults, are detailed in Table 2.
Reported Primary Reasons for Not Undergoing Surgery.

Maternal and Family Characteristics
The median maternal age at birth was 30 years (IQR 27-34 years), with 5 mothers older than 35. While 25 mothers received standard prenatal care, the rest had late or intermediate care, resulting in unconfirmed or partial prenatal diagnoses of congenital deformities. Despite this, all 19 patients (73.1%) born after 2005, except 1 who was born in Ecuador, had prenatal diagnoses of their genetic syndromes or congenital defects. This enabled these parents to be aware of their child's conditions and to seek intervention or palliative care during the neonatal period immediately after birth. Six mothers had poorly controlled gestational diabetes or morbid obesity during pregnancy, and 3 reported using various substances early in pregnancy.
Most patients had no family history of congenital abnormalities or genetic syndromes, and all had healthy liveborn siblings. Although 1 patient reported a family history of CL/P, 2 patients reported a family history of genetic conditions involving inheritable pathogenic variants, which had no direct connection to their diagnoses.
Comparison to Surgically Repaired Patients
We compared the cohort of 26 unrepaired patients to a control group of 2070 well-documented patients who underwent CL/P surgical repair at our institution. While there was no significant difference in gender distribution between the groups, the group of unrepaired patients had a significantly higher proportion of combined cleft lip and palate cases (19 patients, 73.1%) compared to the control group (876 patients, 42.3%; P < .005). In addition, as of May 2026, 82 patients in the control group had passed away, with a median age at death of 61.9 years (IQR 31.3-77.5 years), which is significantly higher than the age at death in our 26 patients’ cohort (P < .0001), though below the general American life expectancy of approximately 78 years. Another key finding was that all 26 patients (100%) had either genetic syndromes or severe congenital anomalies, a significant contrast to 24% (n = 496) of those who underwent surgery (P < .0001). Multivariable logistic regression analysis showed that neither gender nor cleft type predicted early mortality prior to surgery. However, a confirmed diagnosis of genetic syndromes or severe congenital anomalies was associated with an extremely elevated risk of early mortality before repairs (OR = ∞; 95% CI: 18.73 to ∞; P < .0001). This infinite OR occurred because, among our patients, no one without a genetic syndrome or severe congenital anomaly experienced early mortality prior to surgical repair.
Discussion
This study addresses a gap in research on a small, vulnerable subset of patients. While early surgical intervention and supportive care are typically successful in treating CL/P, enabling them to achieve normal life expectancy, 16 our findings provide unique information for a population for whom surgical correction is either not possible or not pursued due to early mortality or complex medical conditions. Unlike previous studies that reported overall mortality of all CL/P individuals, regardless of their surgical status, our focus is on the mortality of patients who did not survive to undergo cleft repair. This is because broader analyses of all cleft patients cannot adequately guide surgical decisions for high-risk populations.
Consistent with existing publications reporting overall mortality, our study demonstrated that prerepair mortality is also primarily driven by co-occurring genetic syndromes or severe congenital anomalies, not the cleft itself.1,3,8,11,16–22 The causes of death in our unrepaired patients are similar to those reported in larger population-based studies. For instance, data from the Centers for Disease Control and Prevention showed that among all CL/P patients, trisomy 13 (14.9%), holoprosencephaly (7.2%), and trisomy 18 (3.5%) were common causes of death from 2000 to 2019. 1 Our study reinforced that in patients who did not undergo surgery, mortality is predominantly contributed by trisomy 13 (30.8%), holoprosencephaly (23.1%), and congenital cardiac defects (11.5%). Similarly, in South Korea, congenital cardiac defects (31.69%) and chromosomal abnormalities (30.99%) were reported as leading causes in CL/P from 2006 to 2018. 23 In a population-based study in Canada, when mortality was adjusted for the presence of congenital or chromosomal anomalies, there was no significant difference in the risk of death between children with orofacial cleft (OFC) and those without OFC. 3 Only 1 death in our unrepaired patients was directly caused by CL/P-related complications, further confirming that the cleft itself is rarely a main cause of early deaths.
Furthermore, our findings on cleft types and mortality align with existing literature on overall mortality. The specific type of cleft plays a role, with some evidence suggesting that isolated cleft palate or combined cleft lip with palate carry a higher mortality risk than isolated cleft lip, often due to a stronger association with syndromic conditions.2,4,8,10,11 A meta-analysis of 21 CL/P mortality studies showed that patients with isolated cleft lip have lower odds of mortality compared with combined cleft lip and palate (OR = 0.28 [0.14, 0.56], P = .005) or isolated palate (OR = 0.34 [0.24, 0.48], P = .0005). 2 Our cohort supports this finding, with only 1 patient presenting with an isolated cleft lip, while 73.1% had combined cleft lip and palate. Thus, the findings of this study are more applicable to patients with cleft palate (either isolated or combined), who carry a higher risk of early mortality. However, logistic regression did not show that cleft type itself was a significant predictor of early mortality, indicating the underlying anomalies are the dominant factor.
Our findings also provide information on implications for surgical planning for surgeons. Cleft lip repair is typically recommended between 3 and 6 months of age, and cleft palate repair is usually performed later, generally between 6 and 12 months of age, or even up to 18 months at some centers.24–26 However, the median age at death in our unrepaired cohort was only 25 days (IQR 4-125 days). We observed 2 distinct subsets within the unrepaired group: those who did not survive long enough to reach the recommended age for surgery, and those who did but did not undergo repair due to other complex medical considerations. Fifty percent of patients died before 25 days of age and 75% died before 125 days of age (approximately 4 months). Specifically, subgroup calculation demonstrated median ages at death of 29 days (IQR 4.5-116.5 days) in patients with genetic syndromes, 4 days (IQR 2-4.5 days) in those with isolated holoprosencephaly, and 58 days (IQR 27-125 days) in those with isolated severe cardiac deformities. These findings indicate the extremely poor prognosis of holoprosencephaly. Surgical planning for patients with severe cardiac deformities or genetic syndromes may be considered beyond a certain age, but should be carefully balanced with the overall prognosis. When independent of CL/P, the mortality of trisomy 13, trisomy 18, severe congenital cardiac defects, and holoprosencephaly is high, particularly in the neonatal period. Reported median survival for trisomy 13 is 7 to 12.5 days, with 1 year survival of 5% to 20%. For trisomy 18, median survival is 9 to 14.5 days, with 1 year survival of 5% to 13%.27–30 Holoprosencephaly, especially the alobar form, carries a 95% mortality within the first year of life. 31 In population-based cohorts, severe congenital cardiac disease shows a 1 year survival of approximately 75% to 83% in the United States, though mortality remains highest in the first year.32,33 A recent registry study in Malaysia reported the median survival age for patients with severe congenital cardiac defects is 3.7 months. 34 These data support our interpretation that early cleft repair planning is generally deferred for patients with holoprosencephaly or lethal chromosomal diagnoses because of other medical complexities and high mortality rate. For severe cardiac defects, cleft repair should be considered once cardiac status is stabilized and short-term survival beyond approximately 4 months is reasonable in medically complex infants. At this point, the likelihood of survival significantly improves, allowing for a more informed decision on the necessity of surgery, balancing the risks of medical intervention with potential benefits for the child's quality of life. This perspective is supported by Rao et al, who reported that in their hospital in India, about 50% of patients with congenital heart diseases were not taken for cleft surgery due to potential risks. 35 Studies have reported optimistic outcomes of operating on CL/P patients with severe congenital defects.5,22,36,37 For example, data from the Toubat et al in Los Angeles showed that, although congenital cardiac defects co-occurring with CL/P brought substantial complexity to surgical management and increased major morbidity, they did not significantly affect overall mortality. 36 This is also consistent with the positive experience from Appel et al in Texas, who reported that patients with trisomy 13 and 18 can sometimes tolerate surgery without complications. Their findings suggest that intensive medical management combined with surgical intervention may help extend life, although initial survival remains the primary challenge. 5 Notably, these cases involved patients who were considerably older than the typical age for cleft repair, undergoing primary cleft repair at 2 and 7 years of age respectively. Also, in the cohort reported by Toubat et al, only 5 of the 63 patients underwent cardiac surgery after their cleft repair, while the remaining patients received CL/P repair until their cardiac conditions were surgically corrected. The time interval between CL/P repair and cardiac surgery was 976 days (∼2.6 years, IQR 632-3072). 36 All in all, surgical timing should always be individualized according to each patient's medical condition, nutritional status, and developmental readiness. For infants who remain medically stable or treated beyond a certain age, surgical planning for CL/P can proceed with greater confidence regarding their survival and potential benefit from repair.
In our cohort, palliative care was not an uncommon option chosen by patients’ families, with a total of 8 patients undergoing palliative care without considering surgical intervention as per mutual decisions between parents and medical teams. Among the patients who did not opt for palliative care, 3 were waiting to complete their cardiac surgery or kidney transplant to be ready for cleft repairs, and 1 was waiting to reach a safe body weight for surgery. The decision of surgical repair should always be weighed by whether the benefits of surgery (principles of beneficence and autonomy) were worth the risk of harm (principle of nonmaleficence) or futility among these patients.4,5,37–39 Individualized and case-specific surgical planning remains the ultimate and ethical approach.
In order to help families and surgeons make timely, informed decisions and allow enough time to understand and process the situation, the role of prenatal diagnosis is crucial. In our cohort, 73.1% (n = 19) of patients born after 2005 (excluding 1 born in Ecuador) had accurate prenatal diagnoses, despite the quality of their prenatal care. For these cases, mothers over the age of 35 routinely underwent genetic testing, while others received it after abnormalities were detected on prenatal ultrasound. Early identification allowed surgeons to recognize high-risk cases sooner and provided parents with a clearer understanding of available options and realistic expectations. While studies suggest that the quality of prenatal care and education may be a risk factor for the occurrence of CL/P,3,21,40 our findings indicate that even inadequate prenatal visits can still be very useful in detecting genetic syndromes or severe congenital anomalies that could affect surgical planning. In many cases, a single genetic test or prenatal ultrasound can detect potential serious conditions early, helping guide clinical management and family counseling.4,39,41
Studies have reported a higher mortality rate in Black and American Indian individuals compared to White individuals in CL/P.1–3,7 While our study was conducted in the Midwest region, with the majority of our patient population being White, we observed that up to 65% of this cohort reported experiencing limited finances or homelessness. This finding suggested that socioeconomic status might be a contributing factor to the vulnerabilities within these patients, regardless of ethnic background.
The limitations of this retrospective study include challenges in obtaining detailed medical records for patients in the earlier years. Although we identified more patients meeting our inclusion criteria, a lack of documentation before 1970 prevented us from achieving a larger sample size or analyzing mortality trends in this specific population subset over time. Another limitation is the lack of population-based data due to limited national registry resources in the United States. 8 Thus, we believe a single-center retrospective study from a tertiary medical center is valuable. To address this limitation, our ongoing work aims to obtain population data from Olmstead County in Minnesota through an epidemiological project and by building a dedicated registry for CL/P patients seen at our medical center. Third, we did not collect data on patients with trisomy 13, trisomy 18, or severe cardiac or central nervous system defects who did undergo surgical repair. This will be our future goal to analyze what surgical intervention might potentially benefit their life expectancy.
Conclusions
The main takeaway for surgeons from this study is the importance of genetic and prenatal genetic testing to guide surgical management in CL/P. For patients diagnosed with trisomy 13, holoprosencephaly, severe cardiac defects, or trisomy 18, ethical surgical planning is essential. Understanding the genotype–phenotype relationship of genetic testing is helpful. These factors should guide treatment decisions, potentially including waiting until patients with more severe conditions are stabilized to better assess their survival likelihood before moving to surgical intervention.
Footnotes
Ethical Considerations
This study was approved by Mayo Clinic Institutional Review Board.
Informed Consent
Informed consent has been waived by our Institutional Review Board.
Author Contributions
Project conceptualization and direction are attributed to Rou Wan, Elameen A. Adam, Ethylin W. Jabs, Pavel N. Pichurin, Samir Mardini, and Waleed Gibreel. Rou Wan led data collection. Primary methodology, statistical analyses, figure creation, and manuscript preparation were performed by Rou Wan, Elameen A. Adam, and Waleed Gibreel. Ethylin W. Jabs, Pavel N. Pichurin, and Samir Mardini contributed to study design, data interpretation, and manuscript editing and finalization. All authors have read and approved the final submitted manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
This data is not available to the public but may be obtained from the authors upon reasonable request.