
Abstract
Early-onset Marfan Syndrome (eoMFS) is caused by pathogenic variants in the fibrillin-1 (FBN1) gene. Typical clinical findings include a prematurely aged appearance, severe atrioventricular valve dysfunction (mitral/tricuspid valve insufficiency), and skeletal findings—primarily arachnodactyly, multiple joint contractures, and pectus deformity. Craniosynostosis has been reported in rare cases of eoMFS, but not in association with the c.3037G>A, p.Gly1013Arg variant in FBN1. Here we present a case of a 2-month-old male with bilateral lambdoidal and sagittal craniosynostosis diagnosed with eoMFS after genome sequencing identified a de novo pathogenic variant in FBN1, c.3037G>A, p.Gly1013Arg.
Background
Marfan syndrome (MFS) is an autosomal dominant connective tissue disorder caused by pathogenic variants in the FBN1 gene. Roughly 75% of patients with MFS have an affected parent, and approximately 25% have a de novo FBN1 variant. 1 MFS was first described by French pediatrician Dr. Antoine Marfan in 1896 when he observed disproportionately long limbs, hands, and feet in a 5-year-old girl. 2 FBN1-related MFS results in a broad range of phenotypes from mild (one or few body systems affected) to severe or eoMFS where multiple organ systems are impacted. Most often, MFS is characterized by severe orthopedic issues (scoliosis, pectus deformities, and dolichostenomelia), significant ocular issues (ectopia lentis, glaucoma, cataracts, and retinal detachment), and aortic root enlargement with risk of dissection. 3
MFS affects approximately 1 in 5000 patients. With appropriate management, affected individuals have the same life expectancy as the general population. Due to the wide range of phenotypes and clinical manifestations of MFS that emerge with age, diagnosis is often delayed in pediatric patients.
eoMFS is a rare and severe form of MFS characterized by early-onset and rapidly progressive cardiovascular disease, with severe skeletal manifestations such as arachnodactyly, joint contraction in flexion, and a prematurely aged appearance (previously described as a “senile” facial appearance). eoMFS is twice as rare as classical MFS (about 1 in 10 000 vs 1 in 5000), and 92% of cases are due to de novo pathogenic/likely pathogenic variants with more severe phenotypes and poorer prognoses than classical MFS. 4
The FBN1 gene encodes fibrillin-1, a protein that forms microfibrils that provide structural support and elasticity for connective tissue throughout the body. 5 The pathogenic mechanism in MFS involves structural insufficiency and dysregulated growth factor-beta (TGF-β) signaling, specifically an increase in TGF-β activity, both of which contribute to the cardiovascular, skeletal, and ocular manifestations of the disease. 6
Individuals with eoMFS typically harbor pathogenic or likely pathogenic variants within a hotspot region (exons 24-32) often referred to as the “neonatal region” of FBN1. 7 Functional studies demonstrate that variants in this region block wild-type FBN1 assembly, causing more severe structural defects and greater TGF-β dysregulation compared to pathogenic variants seen in classic MFS. 8
Of note, craniosynostosis is an extremely rare association with MFS but has been suggested previously. Miller et al found a de novo pathogenic variant in FBN1, c.8226+5G>A, in a young child with sagittal synostosis and features originally thought to be due to Shprintzen-Goldberg syndrome (SGS). 9 The genetic findings ultimately led to a diagnosis of eoMFS, which had lifelong management implications. Other authors have also suggested the role of early craniofacial development with FBN1.10,11
In this report, we present a case of a 2-month-old male with bilateral lambdoidal, sagittal, and metopic synostosis, notable facial features, and severe mitral valve regurgitation with left-sided heart enlargement. Clinical genome sequencing identified a de novo pathogenic variant in FBN1 noted as c.3037G>A, p.Gly1013Arg. The multisuture craniosynostosis in our case may represent a novel or underreported feature within the eoMFS-related developmental continuum.
Case Presentation
The patient was born at 38 weeks gestation via scheduled Cesarean section to a healthy, non-consanguineous couple after an uncomplicated pregnancy. Birth weight was 3160 g (25th percentile), length 49.5 cm (50th percentile), and head circumference 35.6 cm (50th percentile). Newborn nursery course was notable for transient hyperbilirubinemia and otherwise unremarkable.
The patient first presented to the pediatric craniofacial clinic at 2 months of age. The primary care provider had concerns about the patient's head shape and referred the patient for plagiocephaly evaluation. Examination revealed a parallelogram-shaped head with an anterior right frontal prominence and right occipital flattening, consistent with right positional posterior plagiocephaly. No cranial ridging was initially appreciated, and conservative management was recommended, including tummy time and encouraging more time spent on the left occiput.
The patient returned to the craniofacial clinic at 4 months of age for follow-up.
Physical examination was notable for palpable ridges along multiple sutures and persistent cranial asymmetry. This prompted skull radiography, which raised concern for multisuture craniosynostosis, including the sagittal, coronal, and lambdoid sutures (Figure 1).
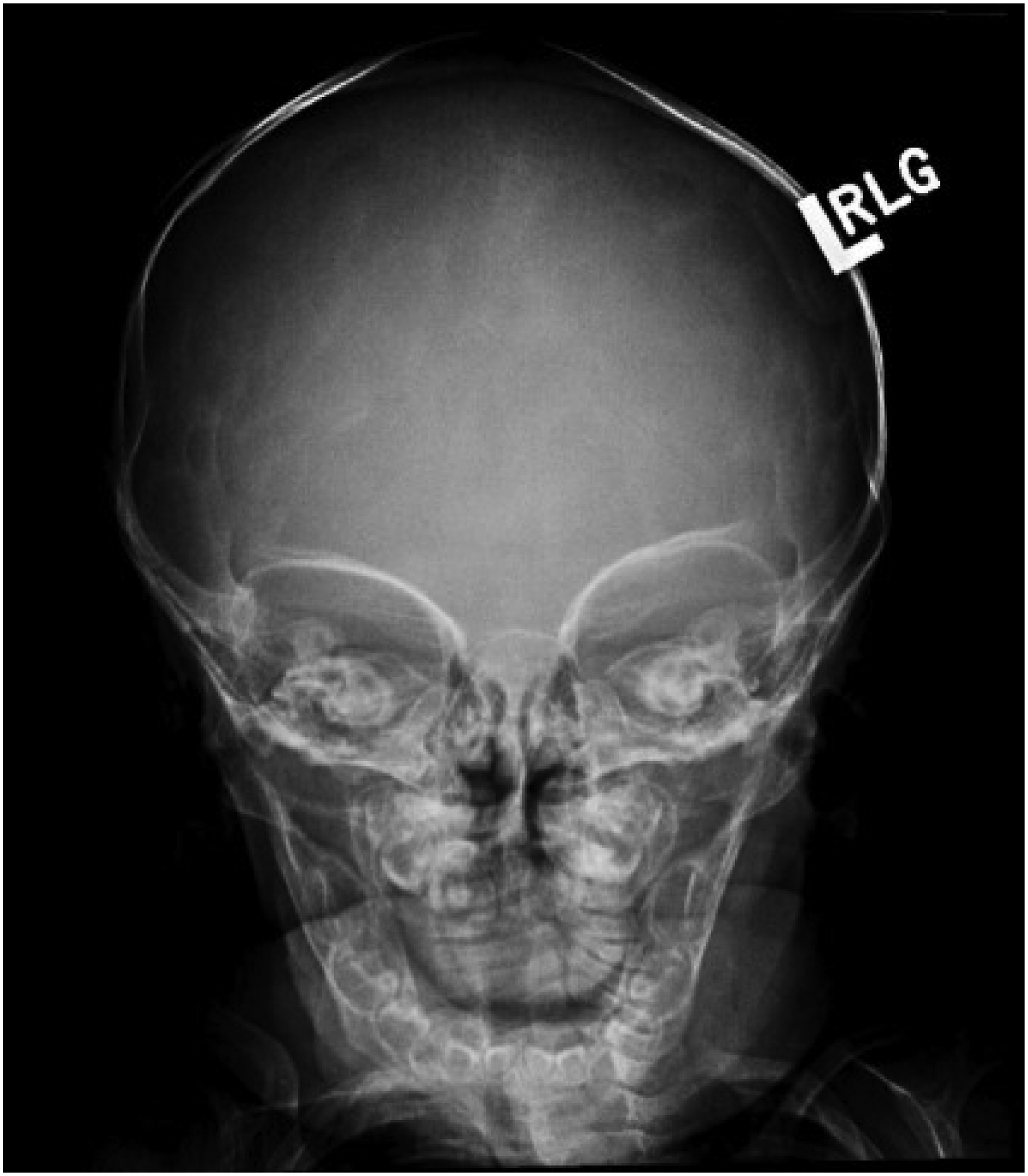
Skull X-ray suggesting possible fusion of the sagittal, coronal, and lambdoid sutures.
CT imaging 1 month later confirmed bilateral lambdoid sutures (Figure 2A) and sagittal suture fusion (Figure 2B). The CT imaging report also noted fusion of the metopic suture. The metopic suture was not commented on in prior imaging and was initially observed at 5 months of age, when fusion would have been developmentally appropriate. No Chiari malformation or elevated intracranial pressure was noted clinically or radiographically.
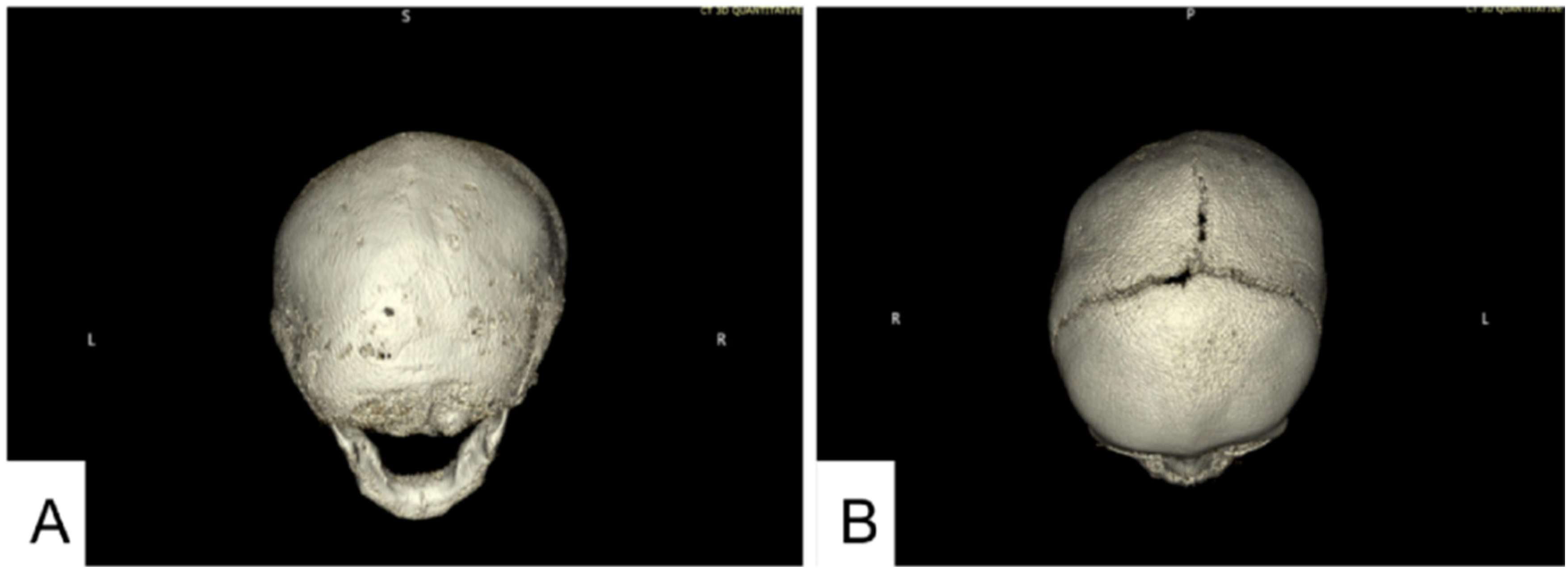
CT done at 5 months of age showing fusion of the metopic, sagittal, and lambdoid sutures. (A) Craniofacial CT 3D reconstruction showing fusion of bilateral lambdoid sutures. The posterior fontanelle was not seen. (B) Craniofacial CT 3D reconstruction showing portions of the superior sagittal suture fused anterior to the coronal sutures and posteriorly. The coronal sutures remain patent, and a residual anterior fontanelle is present. The metopic suture is fused.
Given diagnostic findings of multisuture craniosynostosis, the patient was referred to the craniofacial plastic surgeon and neurosurgeon for cranial vault remodeling surgery planning and medical genetics for consultation, as is the typical course for craniosynostosis in our multidisciplinary pediatric craniofacial clinic.
Meanwhile, around 5 months of age, the primary care provider had noted a cardiac murmur. This prompted referral to a pediatric cardiologist, with evaluations occurring at 8 and 11 months of age, while workup for multisuture craniosynostosis with craniofacial subspecialists was occurring. An echocardiogram revealed severe mitral valve regurgitation due to a floppy, prolapsing mitral valve with annular dilation and left atrial enlargement. Biventricular function remained normal.
Given the absence of symptoms and overall good growth, cardiothoracic surgical intervention was deferred with plans for close monitoring and initiation of afterload reduction therapy. Following discussions between a craniofacial plastic surgeon, a neurosurgeon, and a pediatric cardiologist, cranial vault remodeling surgery was also deferred due to cardiovascular risk concerns.
Genetics physical examination at 11 months old was notable for a long face, frontal bossing, ridging along the lambdoidal and sagittal sutures, closed anterior and posterior fontanelles, downslanting palpebral fissures, malar flattening, a long and thin philtrum, retrognathia with a horizontal chin crease, a high palate, and thick helices (Figure 3). Additional findings included pectus carinatum (Figure 3), joint hypermobility, and striking hand anomalies with arachnodactyly, ulnar deviation of digits, abnormal palmar creases, soft distal digital creases (Figure 4), and long feet and toes.
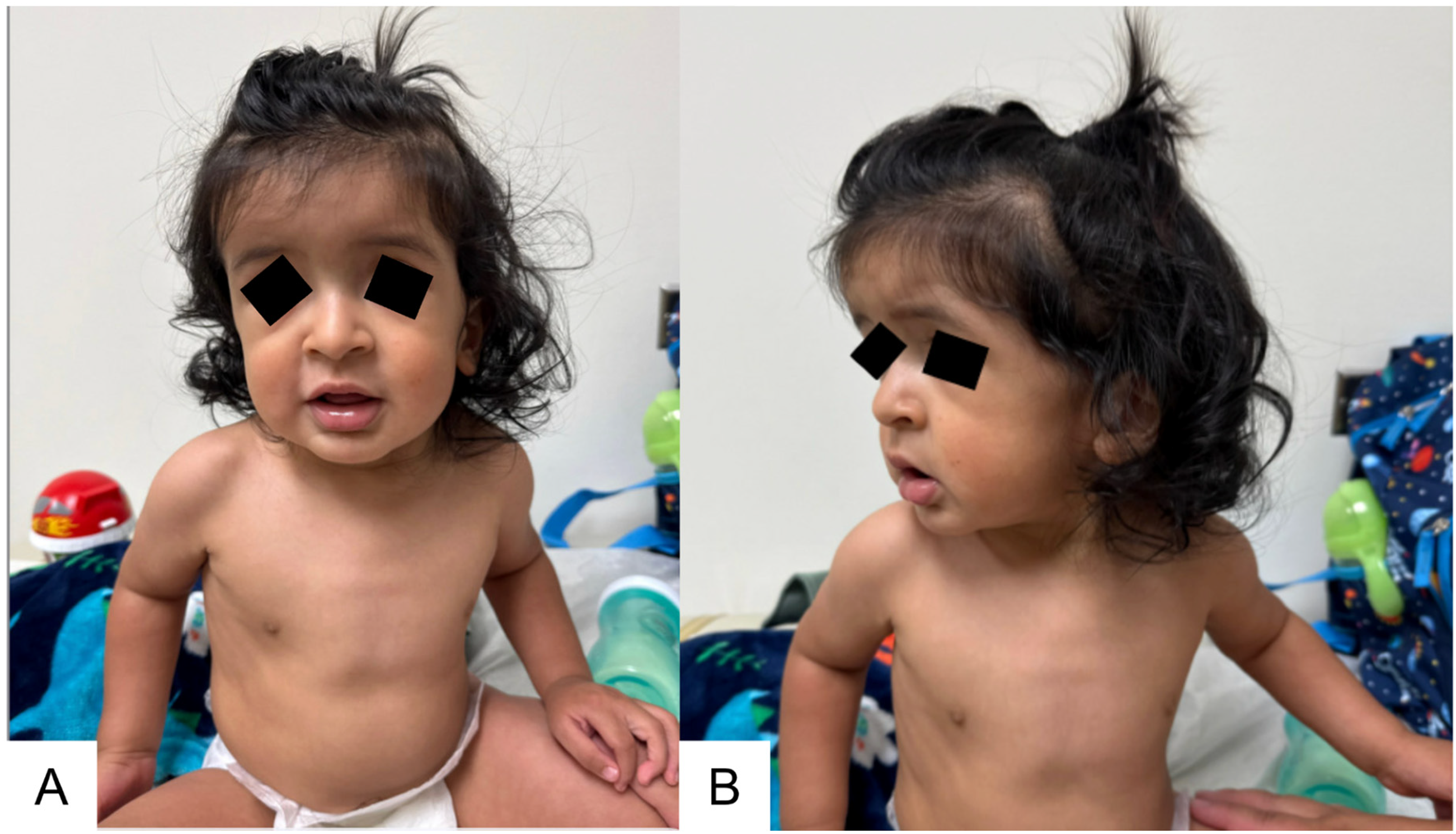
Facial features consistent with eoMFS and additional features secondary to the multisuture craniosynostosis. (A) Frontal photo notable for characteristic “senile” facial appearance, long face, frontal bossing, malar flattening, downslanting palpebral fissures, long and thin philtrum, and pectus carinatum. (B) Side profile view showing frontal bossing, retrognathia with a horizontal chin crease, and thick helices.
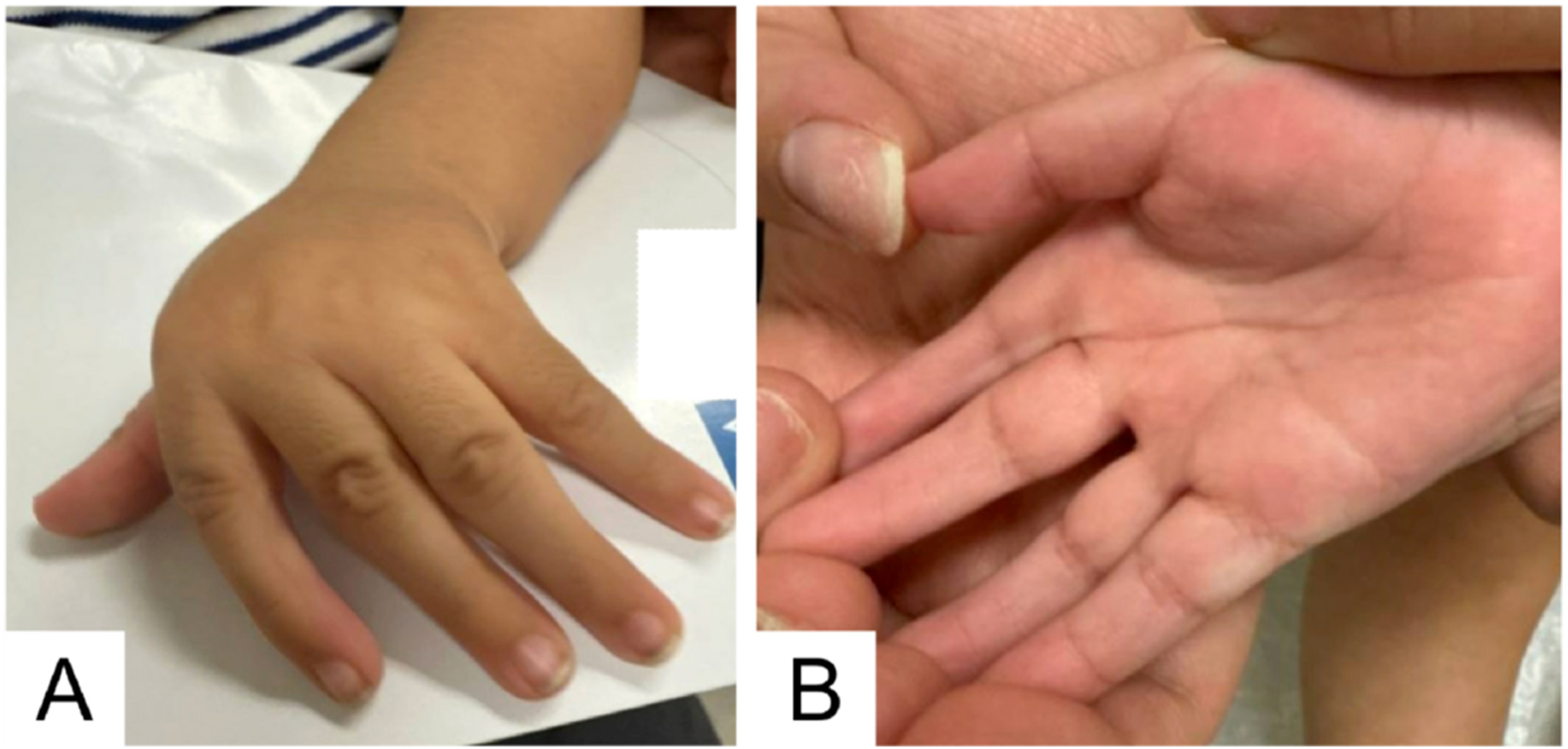
Arachnodactyly and upper limb joint contractures are consistent with the skeletal features of eoMFS. (A) Left hand: Arachnodactyly, ulnar deviation of wrists and digits, fixed flexion contracture at the wrist and metacarpals. (B) Right hand: Arachnodactyly; soft distal digital creases; abnormal palmar creases with longitudinal creases rather than transverse, reflecting the position of the hands with ulnar deviation of digits prenatally and postnatally.
Given the constellation of multisuture craniosynostosis, connective tissue features, notable facial features, and mitral valve disease, the genetics team pursued trio genome sequencing via a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory using a buccal sample for the proband and buccal samples for each parent to be used as comparators. Trio genome sequencing revealed a de novo pathogenic variant in the FBN1 gene: c.3037G>A, p.Gly1013Arg, consistent with a diagnosis of eoMFS.
As per the testing laboratory, variant classification was based on the American College and Medical Genetics and Genomics (ACMG) classification guidelines. Evidence to support this classification included (but was not limited to) variant allele frequency (absent) from approximately 15 000 genomes and 123 000 exomes in the Genome Aggregation Database, the fact that the variant had been observed in multiple unrelated patients with eoMFS in the literature,3,12-16 and de novo inheritance.
Our independent review of this variant took into consideration similar pieces of evidence, in addition to in silico prediction tools (AlphaMissense, for example), which predicted a pathogenic effect on protein function, phenotypic overlap between our patient and those who had been reported to have this variant, and ClinVar ID 177648 entry, which had multiple submissions of this variant as likely pathogenic or pathogenic. 17 The genetics team agreed with the testing laboratory's classification of the variant and provided the diagnosis of eoMFS. All DNA isolation, extraction, quantification methods, bioinformatic analysis, and variant calling methods were performed by the CLIA-certified testing laboratory.
The patient continues to be followed by craniofacial plastic surgery, neurosurgery, medical genetics, and pediatric cardiology. Cranial vault remodeling remains deferred due to the elevated surgical risk from his cardiac condition. He is meeting developmental milestones with no regressions and is monitored closely for signs of intracranial hypertension and cardiac deterioration.
Discussion
MFS is a connective tissue disorder that can affect multiple organ systems including the cardiovascular, ocular, and skeletal systems. 1 Clinical manifestations of MFS may range from mild to severe and show intra- and interfamilial variability, but are highly penetrant. 1 Importantly, the c.3037G>A pathogenic variant found in the patient is located in exon 24 of the FBN1 gene (the “neonatal region”), and has been reported in at least 5 individuals from different ethnic backgrounds with eoMFS.3,12-13,16,18-22 eoMFS is generally a more severe form of MFS that manifests in infancy, characterized by a rapid progression of cardiac and skeletal issues, leading to a poorer prognosis compared to classical MFS. 14 None of the patients with the c.3037G>A, p.Gly1013Arg pathogenic variant had a documented presentation of multisuture craniosynostosis.
The current international diagnostic criteria for MFS are outlined by the Ghent nosology, which was first defined in 1996 and revised in 2010 to emphasize ectopia lentis and aortic root dilation. 4 The Ghent nosology is used to evaluate symptoms of MFS with a total systemic score of 20 and a clinically significant score of 7. When aortic root enlargement and ectopia lentis are present with findings expected of MFS, a diagnosis of MFS can be made. MFS characteristics in other organ systems contribute to the systemic score to outline diagnosis when aortic disease is present, but ectopia lentis is not. 2
Workup for eoMFS is typically pursued when there are notable features on prenatal ultrasound, when there is a diagnosis of tricuspid or mitral valve insufficiencies, or when there is a known family history of MFS.
Multisuture craniosynostosis has multiple known etiologies, many of which involve the TGF-β signaling pathway. Multisuture craniosynostosis is a recurring phenotype in TGF-β pathway-associated connective tissue disorders, specifically Loeys-Dietz syndrome (LDS) 1 and 2 and SGS, but has only been reported in extremely rare cases of MFS and has historically served as a discriminating phenotype in diagnosis.2,19,23
Prior case reports support a potential link between FBN1 and multisuture craniosynostosis. Sood et al was the first to propose that FBN1 expresses as early as the 8-cell stage of human embryogenesis and supports a role in craniofacial and central nervous system involvement. 11 That same paper identified a de novo c.8226+5G>A variant in FBN1 in a patient with sagittal and metopic synostosis. Adès et al described 2 patients with FBN1 variants and cranial involvement: one with MFS and abnormal cranial dura, and another with severe eoMFS with an abnormal skull. 24 Lastly, Miller et al found a de novo pathogenic variant in FBN1, c.8226+5G>A, in a young child with sagittal synostosis. 9
The unifying mechanism appears to be altered TGF-β signaling. Likely pathogenic or pathogenic variants that disrupt this function lead to excessive TGF-β activity, which could theoretically affect cranial suture fusion given TGF-β's critical role in bone formation and remodeling. 25 We therefore theorize that our patient's multisuture craniosynostosis may be explained by the FBN1 pathogenic variant. Whole genome sequencing did not identify any other likely pathogenic or pathogenic variants to explain his multisuture craniosynostosis.
Clinical management for MFS focuses on slowing the progression of aortic root dilation, the most common cause of morbidity and early mortality. 1 Patients receive close cardiac monitoring and multidisciplinary care. In eoMFS, severe valvular deficiencies can result in congestive heart failure in pediatric patients. Valvular surgical repair, valvular replacement, or total heart transplantation are pursued when pharmacotherapy is insufficient. 16 Our case highlights a potentially novel genotype-phenotype correlation and raises the possibility of a broader fibrillinopathy spectrum that includes multisuture craniosynostosis. Recognition of this association could expand the clinical phenotype of MFS and improve diagnostic accuracy in infants presenting with overlapping features such as mitral valve disease and abnormal cranial morphology. This would allow for the early detection and treatment that is critical in a rapidly progressive disease like eoMFS.
Footnotes
Ethical Approval
Our institution does not require ethical approval for reporting case reports or case series.
Informed Consent
Written informed consent was obtained from the patient's legal guardian for publication of this case report and accompanying images.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.