
Abstract
Objective:
This article reviews data encompassing the pharmacology, efficacy, and safety of RSVpreF (Abrysvo®), a subunit vaccine approved to prevent respiratory syncytial virus (RSV) in adults aged ≥60 years, infants via maternal transfer of antibodies, and adults aged 18 to 59 at increased risk of severe disease, including immunocompromised populations.
Data Sources:
A literature review was conducted in PubMed, MEDLINE, National Institutes of Health Clinical Trials Registry, and ClinicalTrials.gov from inception through May 2026, using the search terms Abrysvo, RSVpreF, RSV vaccine, and respiratory syncytial virus. Articles from reference lists were included to identify potentially relevant literature. Guidance documents, including Food and Drug Administration (FDA) briefing documents, package inserts, and recommendations from Advisory Committee on Immunization Practices were included.
Study Selection and Data Extraction:
Data were limited to human clinical studies published in the English language, which evaluated the efficacy, immunogenicity, and safety of RSVpreF.
Data Synthesis:
Eight studies are included describing RSVpreF, consisting of 1 phase I, 2 phase II, 4 phase III, and 1 phase IV clinical study.
Relevance to Patient Care and Clinical Practice in Comparison to Existing Agents:
To protect vulnerable infants, maternal immunization is recommended as a single dose between 32 and 36 weeks of gestation, typically from September through January. Current clinical guidance recommends vaccination for all adults aged ≥75, and those aged 60 to 74 with one or more risk factors for severe RSV disease.
Conclusion:
RSVpreF provides protection against RSV for older adults, high-risk/immunocompromised populations, and infants via maternal transfer. RSVpreF effectively reduces the incidence of severe lower respiratory tract infections, bronchiolitis, and hospitalizations.
Introduction
Respiratory syncytial virus (RSV), an enveloped, negative-sense, single-stranded RNA virus, can result in a significant amount of morbidity and mortality, disproportionately affecting individuals at each end of the age spectrum.1-3 The virus is categorized into 2 main subtypes, RSV type A (RSV-A) and RSV type B (RSV-B), both of which utilize a fusion (F) glycoprotein to fuse viral membranes with host cells. 4 Globally, RSV is the leading cause of lower respiratory tract infections (LRTIs) in children under 2 years of age and is second only to malaria regarding deaths in the first year of life.2,3 In the United States (U.S.), RSV accounts for over 50 000 hospitalizations and 100 to 300 deaths annually in children under 5 years of age. Older adults (>65 years old) face a similarly high burden, with annual hospitalizations ranging from 60 000 to 160 000 and deaths estimated between 6000 and 10 000.
Specific populations are especially susceptible to severe disease, including premature infants, children with congenital heart or lung conditions, and older adults with comorbidities such as chronic obstructive pulmonary disease (COPD) or heart failure. While individuals greater than 60 years of age face an increased risk of hospitalization and mortality from RSV, data suggest that the burden of severe disease extends to younger cohorts with underlying health conditions. 5 Notably, 45% to 63% of adults hospitalized with RSV have underlying cardiovascular disease, and hospitalized adults aged >50 years with RSV are at a significantly increased risk of experiencing an acute cardiac event, particularly if they have existing cardiac conditions. 6 Reflecting these risks, updated Centers for Disease Control and Prevention (CDC) recommendations have expanded to include a single dose of the RSV vaccine for all adults aged 75 and older, as well as those aged 50 to 74 who are at increased risk for severe illness due to chronic medical conditions. These high-risk factors include chronic cardiovascular, pulmonary, renal, or hepatic disorders, as well as diabetes mellitus and impaired immune systems. While RSVpreF is U.S. Food and Drug Administration (FDA)-approved for all adults aged ≥60 and for adults aged 18 to 59 at increased risk, clinical prioritization is now increasingly focused on adults aged 50 and older whose comorbidities significantly elevate their risk for severe respiratory tract disease.
Transmission rates of RSV in the U.S. follow a distinct seasonal and annual pattern, with rates typically highest in December and January. 7 It is believed that the epidemiological trends were significantly disrupted by the COVID pandemic, as nonpharmaceutical interventions such as masking and social distancing resulted in reduced viral RSV circulation and greater fluctuations between RSV-A and RSV-B subtypes. 8 However, recent surveillance data show that RSV patterns appear to be returning to prepandemic norms. 9 This seasonality directly informs the timing of clinical interventions; for example, the Advisory Committee on Immunization Practices (ACIP) recommends that maternal immunization with RSVpreF occurs as a one-time seasonal dose administered from September through January in most of the continental U.S. This specific window is designed to ensure that infants receive peak passive protection during the months of highest viral prevalence. 10
Managing RSV involves a dual approach consisting of both prevention and treatment. Prevention strategies include avoidance of RSV exposure as well as passive immunization via monoclonal antibodies, and active immunization through vaccines. Currently, 3 vaccines are available for the prevention of RSV: the subunit vaccines RSVpreF (Abrysvo®) and RSVpreF3 (Arexvy®), and the messenger ribonucleic acid vaccine mRNA-1345 (mRESVIA®). 4 Among these vaccines, RSVpreF is uniquely positioned for use as the only vaccine indicated for use in pregnant individuals to provide passive protection to infants. 10 This maternal immunization strategy offers a distinct approach compared to direct administration of passive antibodies, such as nirsevimab, to the newborn. By triggering the production of maternal antibodies that are transferred across the placenta, RSVpreF ensures the infant is protected from the moment of birth for up to 6 months, potentially avoiding the need for the infant to receive a separate injection. While these preventive options are critical for reducing the incidence of severe LRTI, clinical management for those already infected typically involves addressing symptoms of bronchiolitis and pneumonia, which are the most common complications in infants and the elderly.11,12
Data Sources
A literature search was conducted in PubMed (inception up to February 2026), MEDLINE, and National Institutes of Health Clinical Trials Registry utilizing the search terms Abrysvo, RSVpreF, RSV vaccine, and respiratory syncytial virus. The review incorporated regulatory and clinical guidance documents, including FDA briefing documents, official package inserts, and recommendations from the ACIP, to ensure the findings align with current U.S. regulatory standards and clinical practice.
Clinical Pharmacology
Mechanism of Action
RSVpreF is a bivalent recombinant vaccine that induces an immune response against the fusion (F) glycoprotein located on the surface of RSV. This glycoprotein is a critical component of the virus’s life cycle, as it is required, for RSV’s viral membranes to fuse with and enter host cells. 13 This is essential for the virus’s life cycle because by fusing to the host cells, it allows the virus to unlock the cell, allowing the virus to enter. By inducing an immune response against this protein on RSV, the RSVpreF prevents the virus from successfully invading the host respiratory cells. 13
Immunogenicity and Immune Response
Clinical data show that a single 120 µg dose is highly effective at stimulating the immune system. 14 In human challenge studies involving healthy adults, vaccine recipients experienced a 20.5-fold increase in neutralizing titers (NTs) against RSV-A by day 28. 9 This robust immune response led to an 86.7% vaccine efficacy against symptomatic RSV infection and significantly reduced both the duration of viral shedding and the peak viral loads in those who did become infected. 14
Maternal-Fetal Pharmacology
A unique aspect of RSVpreF’s pharmacology is its application in maternal immunization to protect infants. In the phase IIb clinical study in pregnant women between 24 and 36 weeks of gestation, the vaccine triggered the production of maternal antibodies when administered. 15 Although the clinical study originally demonstrated that the vaccine triggers a robust immune response when administered between 24 and 36 weeks of gestation, current CDC and ACIP recommendations specify that the vaccine should be administered during a narrower window of 32 to 36 weeks of pregnancy. 15 This recommendation applies to individuals who are expected to deliver during the RSV season (typically September-January) and who have not previously received an RSV vaccine. Specifying this 32- to 36-week window in clinical practice is essential to ensure patient safety and to avoid potential administration errors. When administered during this optimal timeframe, maternal antibodies are transferred across the placenta to the fetus, providing the infant with passive immunity that significantly reduces the risk of severe LRTI for up to 6 months (180 days) after birth.16,17
The aim of this review is to assess the available evidence on the efficacy, immunogenicity, and safety of RSVpreF.
Study Selection and Data Extraction
All relevant English-language studies, or studies that could be appropriately translated into English, containing the pharmacology, pharmacokinetics, safety, and efficacy of RSVpreF were selected for review. Studies currently being conducted were identified through ClinicalTrials.gov using search terms “Abrysvo” and “RSVpreF,” which yielded 13 results of completed, active, or recruiting studies. Studies that did not include RSVpreF by Pfizer were excluded. In addition, reference lists of articles were reviewed to identify potentially relevant articles, conference abstracts, or posters.
Clinical Studies
Schmoele-Thoma et al 18 conducted a phase IIa (NCT04785612), randomized, placebo-controlled, double-blinded study investigating the safety, immunogenicity, and efficacy of RSVpreF for the prevention of symptomatic RSV in 70 healthy adults between ages of 18 and 50. Subjects were randomized in a 1:1 manner to receive either a single 120 μg dose of RSVpreF vaccine or a placebo. Twenty-eight days after the injection, all participants were intranasally inoculated with the RSV-A Memphis 37b challenge strain and monitored for 12 days to evaluate the safety, immunogenicity, and efficacy. 18 The study’s primary endpoints included symptomatic RSV infection confirmed via reverse-transcriptase quantitative polymerase chain reaction or viral culture, along with associated clinical symptoms (cough, sore throat, or fever) observed over consecutive days. The study reported an 86.7% vaccine efficacy against symptomatic RSV infection confirmed by detectable viral RNA on at least 2 consecutive days and 100% efficacy for cases confirmed through viral culture. The vaccine group demonstrated a significant reduction in both the duration of viral shedding and peak viral loads compared with the placebo group. Immune response analysis showed that by day 28, vaccine recipients experienced a 20.5-fold increase in NTs against RSV A, whereas the placebo group saw only a 1.1-fold increase. Adverse events (AEs) were primarily mild to moderate, with no serious safety concerns reported. Within the 28 days post-injection, 34% of vaccine recipients and 29% of placebo participants reported AEs, with only one case of enlarged lymph nodes that was determined to be vaccine-related, which was resolved by day 53. Two participants withdrew during the study, but investigators determined that their reasons were due to AEs that were unrelated to the study agent (a positive COVID-19 test and a prolonged QT interval). Overall, there were no immediate or severe safety concerns that were observed during the study (Table 1). 18
Clinical Studies Evaluating the Immunogenicity and Safety of RSVpreF (Abrysvo).
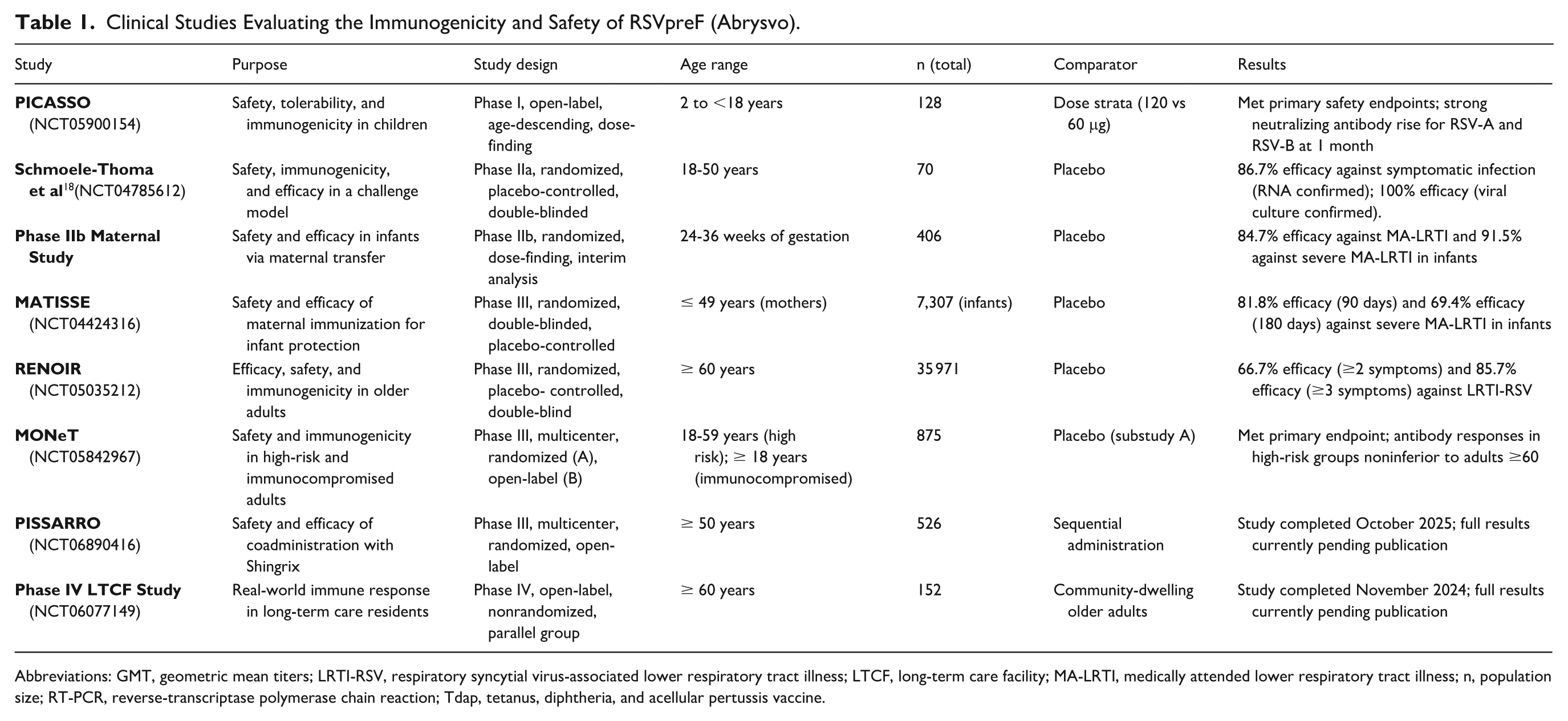
Abbreviations: GMT, geometric mean titers; LRTI-RSV, respiratory syncytial virus-associated lower respiratory tract illness; LTCF, long-term care facility; MA-LRTI, medically attended lower respiratory tract illness; n, population size; RT-PCR, reverse-transcriptase polymerase chain reaction; Tdap, tetanus, diphtheria, and acellular pertussis vaccine.
The phase IIb study of RSVpreF included a single dose of 120 or 240 μg administered to pregnant women between 24 and 36 weeks of gestation. 14 Participants were randomized 1:1:1:1:1 to 120 μg, 240 μg, with or without aluminum hydroxide, or to placebo. A prespecified interim analysis was conducted and all 406 pregnant participants were included related to the safety analyses. The most commonly reported AE was mild-to-moderate pain at the injection site, which occurred more frequently in the group of participants who received the vaccine containing aluminum hydroxide. Serious adverse events (SAEs) reported were considered unrelated to the vaccination with most also unrelated to pregnancy. 14 Furthermore, investigators determined that none of the SAEs reported in the observation phase were related to maternal vaccination. Overall, 3 of the 405 infants in the RSVpreF groups and 5 of the 103 infants in the placebo group were reported to have medically attended RSV-associated LRTI. The observed efficacies against medically attended RSV-associated LRTI and severe RSV-associated LRTI were 84.7% and 91.5%, respectively. 16 The results from this study led to the FDA breakthrough therapy designation for infants up to 6 months of age in preventing RSV-associated lower respiratory tract disease (LRTD). 17
MATISSE (NCT04424316), a phase III randomized, double-blinded, placebo-controlled study evaluated the safety and efficacy of a single 120 μg intramuscular injection of RSVpreF compared with placebo. 15 Healthy women aged ≤49 years who were between 24 and 36 weeks of gestation were assigned to receive RSVpreF or placebo in a 1:1 ratio. Ultimately, infants born to healthy women vaccinated during pregnancy were assessed based on the percentage reduction in the incidence of medically attended respiratory infections due to RSV through 180 days of life and pregnant women were also evaluated for safety and immunogenicity outcomes. 19 A total of 7307 infants (3660 RSVpreF and 3647 placebo) were included in the efficacy population. The primary endpoint of severe RSV-associated medically attended lower respiratory tract illness (MA-LRTI) was significantly reduced in the infants that were born to RSVpreF-vaccinated mothers. Vaccine efficacy against severe RSV-associated LRTI was 81.8% (at 90 days after birth) and 69.4% (at 180 days after birth), which occurred 19 times in the vaccine group compared with 62 times in the placebo group. The secondary endpoint of RSV-associated MA-LRTI of any severity did not meet the criteria for statistical significance, however, LRTI occurred in 24 infants in the vaccine group compared with 56 infants in the placebo group at 90 days after birth, and 57 infants and 117 infants at 180 days, respectively. The most common AE reported was injection-site pain, which was higher in the vaccine group (40.6%) compared with the placebo group (10.1%). 15 Overall, RSVpreF was successful in reducing the number of severe RSV-associated MA-LRTI in infants when the mother was vaccinated during pregnancy while also showing a favorable safety profile. The RSVpreF vaccine was recommended to prevent RSV in infants up to 6 months of age via maternal immunization in the third trimester by the FDA Vaccines and Related Biological Products Advisory Committee. 20
RENOIR (NCT05035212), a phase III, randomized, placebo-controlled study evaluated the efficacy, safety, and immunogenicity of RSVpreF in preventing RSV-associated LRTD in adults 60 years of age or older. The study enrolled a total of 35 971 participants, with 34 284 individuals at interim analysis randomized in a 1:1 ratio to receive either a single 120 μg dose of the RSVpreF vaccine or a placebo. 21 The primary objective was to assess the safety and efficacy in preventing reverse-transcriptase polymerase chain reaction (RT-PCR) assay confirmed LRTI-RSV infection defined by the presence of at least 2 or 3 signs or symptoms lasting more than 1 day, with the primary outcome measure of the number of first episodes of LRTI-RSV cases in the first season beginning 15 days after administration. Interim analysis revealed a vaccine efficacy of 66.7% against LRTI-RSV with at least 2 signs or symptoms on or after day 15 (11 cases in the RSVpreF group vs 33 in the placebo group). 20 For more severe disease involving 3 or more signs or symptoms, vaccine efficacy rose to 85.7% (2 cases in the RSVpreF group vs 14 cases in the placebo group). 20 Overall, the study reported a total of 70 cases of LRTI-RSV during the first season (22 cases in the vaccine group and 58 cases in the placebo group), correlating with a seasonal vaccine efficacy of 62.1%. While the case numbers at the interim stage were small, the study successfully met the prespecified criteria for its primary endpoints. 22 The study design also included 3 substudies, which intended to evaluate participants who are revaccinated with RSVpreF 2 years after their first dose (substudy A), revaccination with RSVpreF 1 year after their initial dose (substudy B), or revaccination with RSVpreF 3 or 4 years after their initial dose (substudy C). 21 These ongoing individual substudies aim to answer critical questions regarding how long the protection against RSV lasts and the potential need for periodic boosters. The individual substudies with redosing of the vaccine will provide additional information about efficacy in the future.
MONeT (NCT05842967), a phase III multicenter clinical study assessed the safety, tolerability, and immunogenicity of RSVpreF in adults at high risk of severe RSV disease. 23 The study included 2 substudies. Substudy A was a randomized, double-blind, placebo-controlled study that evaluated RSVpreF in adults 18 to 59 years of age with specific chronic medical conditions, such as chronic pulmonary (including asthma), cardiovascular (excluding isolated hypertension), renal, hepatic, neurologic, hematologic, or metabolic disorders (including diabetes mellitus) that were not immunocompromised. Substudy A enrolled 675 individuals, with participants receiving either one 120 μg dose of RSVpreF or placebo in a 2:1 ratio. The primary outcome measures for immunogenicity looked at neutralizing antibody geometric mean titers (GMTs) against RSV-A and RSV-B at 1 month postvaccination, geometric mean fold rise (GMFR) from baseline to one month, and seroresponse rates (SRR) defined as ≥4-fold rise in neutralizing antibodies from baseline. The study met the primary endpoint, demonstrating that a single dose of RSVpreF in younger high-risk groups generated neutralizing antibody responses noninferior to those seen in adults ≥60 years of age in previous studies. 23 Substudy B was a single-arm, open-label study consisting of immunocompromised adults aged ≥18 years, of which approximately half of participants were ≥60 years of age. Substudy B enrolled 200 participants in which these participants all received two 120 μg doses, one month apart. The primary outcome measures for safety and tolerability were local reactions (pain, swelling, and redness) within 7 days, systemic events (fever, fatigue, headache, nausea, muscle or joint pain), AEs through 1 month postvaccination, SAEs through 6 to 7 months, newly diagnosed chronic medical conditions, and medically attended AEs. The goal of this study was to determine whether 2 doses are needed for RSV protection in immunocompromised adults, a population that is often excluded from vaccine studies. It was determined that RSVpreF was well tolerated with no new safety issues, only those consistent with expectations for this population. Furthermore, RSVpreF vaccine was found to produce clinically meaningful strong neutralizing antibody titers against RSV-A and RSV-B after just one dose, although receiving the second dose would offer further benefit in boosting antibody levels. 20
A phase IV (NCT06077149) open-label, nonrandomized, parallel-group immunogenicity study aimed to provide data on the immune response of adults over 60 years of age living in a long-term care facility (LTCF) receiving the Pfizer RSV vaccine or the GSK RSV vaccine compared with their same age counterparts living independently in the community. 24 The study enrolled 152 adult participants ≥60 years who were either residents of LTCFs or community-dwelling older adults with a broad inclusion criteria to reflect the real-world populations. The key exclusion criteria included those with immunosuppressive conditions, recent vaccines within 14 days, recent RSV infections within 2 months prior to enrollment, and recent blood products or immunoglobulin (Ig) within 60 days before RSV vaccination. Participants had a baseline blood draw, vaccination between November and December 2023 with follow-up blood draws at 28 to 42 days, and monitoring for intercurrent illness and RSV testing results. Primary outcomes measured GMFR of RSV-A (Fa protein), RSV-B (Fb protein) strains, and RSV-A2 strain (neutralizing antibody fold rise against RSV-A2) at 30 days postvaccination. Secondary outcomes measure the functional antibody quality using the ratio of RSV-A2 NT fold rise to Fa IgG binding titers and will assess the proportion of functional versus binding antibodies. This ratio will help determine whether the antibodies produced against the RSV are functional (neutralize the virus) or nonfunctional (binding but not protective); higher ratio suggests higher-quality immune response. 23 Although the study has been completed as of November 2024, full results of this study are currently not yet available.
PISSARRO (NCT06890416), a phase III multicenter, interventional, open-label, randomized, parallel-group study aimed to determine whether the RSVpreF and recombinant zoster vaccine (RZV) could be safely coadministered and whether immune responses to both vaccines would remain effective when given together. The study enrolled 526 adults ≥50 years who received the RSVpreF vaccine and RZV together (coadministration group) or in a staggered schedule (sequential administration group), depending on their assignment. Participants receiving the sequential administration group were to receive RSVpreF at one visit, then RZV at a later visit, allowing the comparison of safety profiles, immune responses, and reactogenicity patterns. 25 Primary objectives for safety and tolerability were to evaluate participants for local reactions (pain, redness, and swelling), systemic symptoms (fever, fatigue, headache, and muscle aches), AEs, or SAEs. Primary objectives for immunogenicity were to measure immune responses to RSVpreF (neutralizing antibodies to RSV-A and RSV-B) and RZV (glycoprotein E-specific antibodies). Secondary objectives compared GMTs and fold rises for both vaccines between the coadministration and noncoadministration groups, assessing the durability of the immune responses (how long the antibody responses last after vaccination), and evaluating for safety over the full follow-up period. The study will be important in determining whether it is safe and immunogenic for adults ≥50 years of age to be given both RSVpreF vaccine and RZV together, reducing clinic visits, improving vaccination rates, while also simplifying preventive care especially for older adults with multiple chronic conditions. 25 Although the study has been completed as of October 2025, full results of this study are not available at the time of this writing.
Ongoing Research
PICASSO (NCT05900154), a phase I, open-label, age-descending, dose-finding study investigated the safety, tolerability, and immunogenicity of RSVpreF in approximately 128 RSV-seropositive children between the ages of 2 and <18 years. Subjects were divided into 2 age strata: children 5 to <18 years of age (including healthy children and those with high-risk chronic medical conditions) and children 2 to <5 years of age, to either receive a single 120 μg (age 5 to <18) or 60 μg dose (age 2 to <5) of the vaccine. 26 Following a single intramuscular injection, participants were followed for 6 months, with an Internal Review Committee assessing safety data from the older cohort before administration proceeded to the younger age group. The study’s primary endpoints focused on the safety and tolerability of RSVpreF at each dose level, specifically measuring prompted local reactions (pain, redness, and swelling) and systemic events (fever, fatigue, and headache) recorded via an electronic diary for 7 days postvaccination, along with collection of AEs and SAEs. 26 To mitigate the theoretical risk of vaccine-mediated enhanced respiratory disease (ERD), enrollment was limited to seropositive children, with the younger children requiring a screening serostatus test prior to vaccination. 26 Primary outcomes were similar across age groups and doses with common short-lived local reactions being mild or moderate injection-site pain, redness, and swelling; most frequent systemic reactions were mild-to-moderate fatigue, headache, fever, muscle aches, and gastrointestinal symptoms that generally resolved without intervention. Secondary endpoints focus on the immune response, measuring RSV-A and RSV-B NTs, GMTs, and GMFRs at 1 month postvaccination. 26 The study found significant fold rise in titers at 1 month postvaccination, with both doses (60 and 120 μg) producing strong immunogenicity, however, the 120 μg dose produced even higher titers. The study also characterizes cell-mediated immune responses by monitoring RSV F antigen-specific CD4+ T cells to assess the Th1/Th2 cytokine balance. 26 The study showed clear RSV-neutralizing antibody increases after vaccination with responses showing a balanced Th1/Th2 profile. Safety oversight included the implementation of stopping rules for SAEs or grade 4 reactions, and potential risks identified including Guillen-Barre syndrome (GBS) and atrial fibrillation, which were observed at low frequencies in prior adult studies. 26
Safety
No clinically relevant SAEs have been seen with RSVpreF. Injection-site reactions are the most common AE seen with RSVpreF with approximately 40% of individuals experiencing reactions compared with roughly 10% in placebo groups. Other common AEs are typically mild and transient, including arthralgia, fatigue, fever, headache, nausea, and myalgia. In specific studies (phase IIa challenge study), enlarged lymph nodes was the only event deemed vaccine-related. Maternal studies indicated that SAEs in both mothers and infants were largely unrelated to the vaccination. Reflecting regulatory caution, the FDA has required a warning in the prescribing information for RSVpreF regarding the potential of GBS. While cases of GBS and other inflammatory neurologic events were observed at low frequencies in prior studies, they are being actively monitored in Pfizer’s postsurveillance plans. These plans also include the monitoring of atrial fibrillation, syncope, anaphylactoid/hypersensitivity reactions, acute illness, and bleeding disorders. 22
Data regarding coadministration with other vaccines are limited, but currently no restrictions exist with regards to concomitant use. A phase IIb study (NCT04071158) showed a statistically significant reduction of acellular pertussis antibody titers when RSVpreF was coadministered in nonpregnant individuals with the Tdap vaccine compared with Tdap administration alone, however, the clinical relevance is unknown. 27 Despite reduced pertussis titers, there are no current restrictions on coadministration.
Dosage/Administration
The RSVpreF vaccine must be reconstituted prior to administration. RSVpreF may be supplied as a vial of lyophilized antigen plus a “Sterile Water Diluent Component” held within either (1) a prefilled syringe or (2) a vial. Sterile water is not recommended for reconstitution. Following reconstitution, it is administered as a 0.5 mL intramuscular injection. Currently, there are no contraindications to administration aside from those with anaphylaxis to any of RSVpreF’s ingredients. However, of note, one study did observe a decrease in acellular pertussis antigen titers when RSVpreF was given at the same time as a Tdap vaccine, although the clinical impact of this interaction remains unclear. 28
Relevance to Patient Care and Clinical Practice in Comparison to Existing Agents
Vaccination against RSV provides critical protection to individuals while significantly reducing the clinical and economic burden the virus places on the health care system. Rather than focusing on herd immunity, the primary objective of RSV vaccination is the mitigation of severe disease outcomes, such as hospitalizations and deaths that disproportionately affect those at each end of the age spectrum.29,30 In the U.S., RSV is responsible for over 50 000 hospitalizations in children under 5 and up to 160 000 hospitalizations in adults aged ≥60 annually. By preventing these severe clinical complications, the economic impact of widespread vaccination is substantial, with billions of dollars saved in health care costs. 31 This significant reduction in health care burden demonstrates that the return on investment in vaccination far outweighs its cost. 32
Current clinical guidelines for RSV vaccination have evolved; while initially framed as a shared clinical decision-making between providers and patients, vaccination recommendations now differ slightly between the FDA-approved labeling for this vaccine and ACIP.31,33,34 As of May 2026, the FDA label for RSVpreF has 3 groups indicated for the vaccine: individuals 60 years of age and older, pregnant individuals at 32 through 36 weeks of gestational age, and individuals 18 through 59 years of age who are at increased risk for LRTD caused by RSV. 24 At the time of this writing, there are 2 populations in which ACIP recommends RSV vaccination: women who are pregnant at 32 through 36 weeks of gestation from September through January in most of the continental U.S. and unvaccinated adults aged 75 years or older. According to ACIP, individuals aged 50 to 74 years fall under a “Special situations” category, which states those who are unvaccinated and at increased risk of severe RSV disease should receive the vaccine. 34 These individuals with specific risk factors include those with cardiovascular disease (eg, coronary artery disease and heart failure), chronic lung conditions (eg, asthma or COPD), diabetes, or impaired immune systems. 33 Of note, 45% to 63% of adults hospitalized with RSV have underlying cardiovascular disease, 32 suggesting that this population may particularly benefit from vaccination. However, individualized conversations allow providers to address patient concerns and emphasize that the vaccine provides up to 85.7% efficacy against severe LRTI, therefore enhancing vaccine uptake in vulnerable populations.
To protect infants from RSV-associated LRTI, maternal immunization with RSVpreF is approved for use during pregnancy to be administered between 32 and 36 weeks of gestation. However, clinicians should be cognizant of certain vaccination errors that have been reported with RSVpreF. Infants and young children have received the vaccine erroneously instead of nirsevimab, a monoclonal antibody used for RSV prevention. 35 In some cases, GSK’s RSVpreF3 vaccine (Arexvy) has been mistakenly administered to pregnant women. 36 Despite these concerns, ACIP recommends seasonal administration of RSVpreF for pregnant persons as a one-time dose at 32 to 36 weeks of gestation from September through January in most of the continental U.S. 36 Ultimately, clinicians must maintain strict vigilance to avoid administration errors, such as the accidental vaccination of infants instead of providing monoclonal antibodies or the use of incorrect RSV vaccine brands in pregnant patients.
Personalized discussions that balance the risks of vaccination with its preventive benefits hold the potential to significantly enhance vaccine uptake, particularly among high-risk populations.37,38 As of May 2026, a single lifetime dose of the vaccine is recommended for all individuals aged ≥75, as well as patients aged 60-74 with one or more risk factors for severe RSV disease, and in pregnant females between 32 and 36 weeks of gestation from September through January in most of the continental U.S.
For individuals with subsequent pregnancies, it is important to note that current ACIP and American College of Obstetricians and Gynecologists (ACOG) guidance recommends the maternal RSV vaccine as a one-time dose. 38 Unlike the Tdap vaccine, which is administered during every pregnancy to ensure optimal antibody transfer, the RSV vaccine is currently not recommended for repeat administration in subsequent pregnancies if a dose was received previously. Therefore, if a pregnant individual received RSVpreF during a previous pregnancy, they should not receive a second dose in a subsequent pregnancy, even if that pregnancy occurs in a later RSV season. Clinicians should verify a patient’s vaccination history to avoid unnecessary redosing and ensure compliance with these specific clinical guidelines.
Postmarketing Surveillance and Real-World Evidence
Following the regulatory approval and initial implementation of RSVpreF, research has expanded to evaluate its effectiveness in broader, “real-world” clinical settings. A notable example is the phase IV LTCF study (NCT06077149), 23 which is designed to assess the immune response of adults aged ≥60 years living in LTCFs compared with community-dwelling adults. This study is particularly significant as it includes a more frail population often excluded from the initial prospective studies used for licensing.
While the full results from these large-scale postmarketing studies were pending at the time of this review, they are essential for determining if the high clinical efficacy observed in the RENOIR study (up to 85.7% against severe disease) translates into a significant reduction in the 60 000 to 160 000 annual hospitalizations among older adults. One of the limitations of this review is the inclusion of prospective studies and the lack of real-world data. Active postmarketing surveillance is ongoing to monitor the long-term safety profile and the durability of the immune response in the general population, ensuring that the clinical benefits identified in prospective studies are maintained in routine practice. Until these data are collated and analyzed potential AEs may be underreported or unknown.
Future Directions
While the current data for RSVpreF are promising, several areas require ongoing investigation to optimize clinical use. A primary focus is the duration of immunity and the specific time frame in which protection from RSVpreF might wane, and if revaccination will be needed as these questions have not been answered to date. While it is established that the vaccine provides significant protection during the initial RSV season, the long-term persistence of the immune response has not been fully studied. Current research, including ongoing substudies of the RENOIR clinical study, is evaluating the impact of redosing at intervals of 1, 2, 3, and 4 years to determine how long protection lasts and whether periodic boosters will be necessary. In the maternal population, current ACIP guidelines recommend the vaccine as a one-time seasonal dose. However, further research is needed to determine the safety and necessity of revaccination in subsequent pregnancies, a practice that is currently not advised. Expanding protection to the pediatric population is also a key area of study. The PICASSO phase I study recently investigated the safety and immunogenicity of RSVpreF in children aged 2 to <18 years, finding that both the 60 and 120 µg doses were well tolerated and produced strong neutralizing antibody responses with a balanced Th1/Th2 profile. These findings provide a foundation for future larger-scale studies in children. 17 Finally, there are currently no official restrictions regarding the concomitant use of RSVpreF with other vaccines, as the available data are considered limited. Further research is being conducted to determine if coadministration of RSVpreF with other vaccine types is appropriate and maintains the optimal effectiveness, as early data have indicated a potential decrease in certain pertussis antibody titers when administered alongside the Tdap vaccine; however, the clinical significance is unknown (ClinicalTrials.gov). 27 Ongoing studies like PISSARRO are investigating coadministration with the RZV to simplify preventive care for older adults. 26 In addition, phase IV studies in LTCF will soon provide real-world data on immune responses in the most frail elderly populations. 24
Conclusion
RSVpreF induces a robust immune response that translates into significant clinical efficacy against RSV for vulnerable populations. It is FDA-approved for pregnant individuals between 32 and 36 weeks of gestation to safeguard infants via maternal immunization, and for adults aged ≥60 years, and for adults aged 18 to 59 years who are at increased risk of LRTD, particularly those with high-risk comorbidities such as cardiovascular disease, chronic lung disease, or impaired immune systems by reducing the incidence and severity of LRTIs. It is also important to note the current ACIP recommendations for vaccinations which include pregnant women who are between 32 and 36 weeks of gestation during the months of September through January in most of the continental U.S. and unvaccinated adults 75 years and older with the additional special situation category for those with one or more risk factors for severe RSV disease and 50 to 74 years of age. RSVpreF reduces the incidence of medically attended and severe RSV-associated LRTI by 69.4% to 91.5% in infants via maternal immunization and up to 85.7% in adults aged ≥60. This vaccine also provides a critical layer of protection for vulnerable populations, including those with impaired immune systems and those with high-risk comorbidities, such as cardiovascular or chronic lung disease. Safety data show a favorable profile with no clinically serious AEs directly attributed to the vaccine in the primary studies; reactions are typically mild and transient, with injection-site pain being the most common. However, clinicians must maintain strict vigilance to avoid administration errors and ensure the selection of the correct RSV preventive for the appropriate patient in addition to staying current with the latest recommendations to ensure provision of optimal patient care. Ongoing research is essential to determine the long-term durability of protection, the potential necessity for periodic redosing, and the clinical impact of coadministration with other adult vaccines.
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.