
Abstract
Background:
Multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB) impose a disproportionate burden on high-endemic countries, with India contributing the largest global share of rifampicin-resistant cases. The National TB Elimination Program (NTEP) introduced longer all-oral regimens to replace injectable-based therapy; however, prospective pharmacovigilance data characterizing the real-world adverse drug reaction (ADR) profile of these regimens from India remain scarce.
Objective:
To prospectively evaluate the incidence, nature, system organ class distribution, and severity of ADRs in patients receiving longer all-oral MDR/XDR-TB regimens under NTEP at a tertiary care centre in North India.
Methods:
A prospective cohort study enrolled microbiologically confirmed MDR/XDR-TB patients initiating longer all-oral regimens between March 2023 and September 2023, with follow-up through March 2025. Adverse drug reactions were actively monitored via structured interviews and clinical assessments. Severity was graded using the modified Hartwig and Siegel scale. Treatment outcomes were classified per WHO definitions. This study adheres to the STROBE reporting guidelines.
Results:
Of 143 registered patients, 102 (71.3%) were enrolled (52 males [51.0%]; mean age 34.7 ± 14.3 years). Favourable outcomes were achieved in 79 patients (77.5%). A total of 158 ADRs were reported, most of which were mild (Hartwig levels 1-2: 81.0%). Gastrointestinal disorders were most common (n = 57; 55.9%), followed by nervous system disorders, mainly linezolid-associated peripheral neuropathy, requiring dose modification or discontinuation in some cases. Other ADRs included clofazimine-related skin pigmentation, linezolid-associated haematological toxicity, and one case of bedaquiline-associated QT prolongation. Linezolid-related haematological toxicity was the most clinically significant ADR (Hartwig level 4). No severe ADRs (Hartwig levels 5-7) or deaths were reported.
Conclusion and Relevance:
Longer all-oral MDR/XDR-TB regimens demonstrated a favourable safety profile under programmatic conditions, with predominantly mild, manageable ADRs and a treatment success rate exceeding the global MDR-TB benchmark. Proactive monitoring for linezolid-induced neurotoxicity and haematological toxicity, and electrocardiographic surveillance during bedaquiline therapy, are essential.
Keywords
Introduction
Tuberculosis (TB) remains one of the leading infectious causes of mortality worldwide, with an estimated 10.8 million incident cases and 1.25 million deaths reported globally in 2023. 1 The advent of drug-resistant strains—multidrug-resistant TB (MDR-TB), defined as resistance to both isoniazid and rifampicin, and extensively drug-resistant TB (XDR-TB), encompassing additional resistance to fluoroquinolones—has substantially undermined global elimination targets. 2 India bears the greatest global TB burden, accounting for an estimated 1.1 million rifampicin-resistant or MDR-TB cases in 2023—approximately 27% of the global total.1,3 The COVID-19 pandemic compounded this challenge by disrupting case notification, treatment initiation, and continuity of care, with consequent implications for resistance amplification. 4 Multi-state surveillance data from India have documented MDR-TB prevalence rates of 3% among new cases and 12% to 17% among previously treated patients, underscoring the magnitude and heterogeneity of drug resistance across the country. 5 Global MDR-TB treatment success rates remain unacceptably low at 56%, with XDR-TB achieving only 39%. 6 These outcomes reflect the clinical complexity of prolonged, polypharmacy regimens, poor tolerability, and significant rates of loss to follow-up. 7
In 2020, India transitioned from the Revised National Tuberculosis Control Program to the National Tuberculosis Elimination Program (NTEP), which incorporated standardized, longer all-oral regimens for MDR/RR-TB—a pivotal shift away from injectable aminoglycoside-based treatment strategies. 8 The programmatic introduction of bedaquiline, a first-in-class diarylquinoline group A agent, facilitated the assembly of fully oral regimens guided by drug susceptibility testing (DST), improving treatment feasibility and eliminating injection-related morbidity. 8 Despite these advances, all-oral regimens incorporate multiple second-line agents—levofloxacin (Lfx), bedaquiline (Bdq), linezolid (Lzd), clofazimine (Cfz), and cycloserine (Cs)—each associated with distinct and potentially serious ADR profiles encompassing neurotoxicity, hematotoxicity, cardiotoxicity, and dermatological reactions. 9 Such reactions may necessitate dose modification, temporary discontinuation, or permanent withdrawal of key agents, with direct implications for treatment success and patient safety.
Although prior studies from high-burden settings have reported ADRs associated with individual second-line antitubercular agents,1,10 prospective, systematically collected pharmacovigilance data from India—particularly from tertiary care settings in North India—remain limited. This study addresses this evidence gap by prospectively characterizing the incidence, organ-system distribution, and severity of ADRs in patients receiving longer all-oral MDR/XDR-TB regimens under NTEP. The findings are intended to provide regulatory-grade real-world safety evidence to inform programmatic pharmacovigilance, optimize clinical monitoring protocols, and support evidence-based policy decisions for drug-resistant TB management.
Method
Study Design and Setting
This prospective observational cohort study was conducted at a tertiary care referral centre for drug-resistant TB in Delhi, India, and is reported in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines. 11 Patients were consecutively enrolled from March 2023 to September 2023, with active follow-up until March 2025 (minimum 18 months per patient). The study centre functions as the principal NTEP implementation site for North India, providing standardized programmatic MDR-TB treatment free of charge.
Participants
Eligible patients were adults (≥18 years) with microbiologically confirmed MDR-TB or XDR-TB who initiated longer all-oral regimens under NTEP. Microbiological confirmation required positive sputum culture with DST by line probe assay (first-line line probe assay [FL-LPA] or second-line line probe assay [SL-LPA]) or conventional DST. Exclusion criteria were: age <18 years; pregnancy or lactation; concurrent conditions expected to confound ADR attribution (eg, preexisting severe peripheral neuropathy); migration from the study catchment area before outcome declaration; refusal to provide written informed consent; or modification of the prescribed regimen within 4 weeks of enrolment.
Patient Recruitment and Eligibility Flow
Between March and September 2023, 143 patients registered at the study site were screened. Fourteen patients (9 pregnant women; 5 minors) were excluded per eligibility criteria. Twelve declined written informed consent. Eight migrated before treatment records could be accessed. Seven declined further participation within 4 weeks of treatment initiation and were lost to follow-up before any ADR data could be documented; these were excluded from analysis. The final analytic cohort comprised 102 patients, all of whom were followed for treatment outcome declaration. Loss to follow-up in course of treatment was classified as an outcome, not as exclusion.
Treatment Regimen
All enrolled patients received the NTEP-recommended longer all-oral regimen of 18 to 20 months of duration, devoid of a distinct intensive phase. 12 For patients weighing >70 kg, the regimen comprised: Lfx 1000 mg daily; Bdq 400 mg daily for 2 weeks, followed by 200 mg 3 times weekly for 22 weeks; Lzd 600 mg daily, tapered to 300 mg at 6 to 8 months to mitigate cumulative toxicity; Cfz 100 mg daily; Cs 750 mg daily; and pyridoxine 100 mg daily as neuroprotection. 12 Doses were weight-adjusted for patients ≤70 kg per programme guidelines. Medicines were dispensed free of charge from the hospital pharmacy under NTEP.
Treatment Monitoring and Outcome Definitions
Culture conversion was assessed at months 4, 5, 6, and 8. Where month 5 culture was unavailable, the month 4 result was substituted. Extended Lzd at 600 mg was continued for up to 2 additional months if months 6 to 7 cultures remained positive, guided by clinical or radiographic response. Culture positivity at month 8 was classified as treatment failure, triggering comprehensive reevaluation including FL-LPA, SL-LPA, and conventional culture-DST. Treatment outcomes were defined according to World Health Organization (WHO) 2022 definitions: cure (documented sputum culture conversion and completion); treatment completion (completion without documented culture conversion); treatment failure (treatment terminated early or permanently switched due to lack of response); lost to follow-up (treatment interrupted for ≥2 consecutive months); and death (any cause during treatment).
Data Collection
Baseline clinical and demographic data were extracted from NTEP treatment cards and supplemented by structured patient interviews using a validated, pretested semi-structured questionnaire. Interviews were conducted in Hindi by trained research pharmacists and lasted 15 to 20 minutes. Clinical assessments included a detailed history of prior antitubercular therapy, substance use, comorbidities, and anthropometric evaluation (height, weight, and body mass index). Baseline investigations included sputum smear and culture with DST, complete blood count, liver and renal function tests, serum electrolytes, fasting blood glucose, urine analysis, HIV serology (postcounselling), chest radiography, and 12-lead electrocardiography (ECG). Ophthalmic assessment (visual acuity and colour vision) was performed at baseline and repeated at 3-monthly intervals given linezolid use. ECG was performed at baseline and at monthly intervals during bedaquiline therapy to monitor QTc.
ADR Assessment
Adverse drug reactions were actively monitored at each scheduled clinical visit (monthly) through standardized clinician review, patient-reported symptoms, and laboratory reevaluation. Adverse drug reactions were documented using MedDRA System Organ Class (SOC) terminology. Severity was graded using the modified Hartwig and Siegel severity scale, categorizing reactions as mild (levels 1-2: does not require treatment modification or is managed symptomatically without dose change), moderate (levels 3-4: requires dose modification, specific treatment, temporary, or permanent drug discontinuation), or severe (levels 5-7: life-threatening, causes permanent disability, or contributes to death). Suspected causal agents were identified by clinical pharmacists based on temporal relationship, known drug toxicity profiles, and clinical presentation.
Statistical Analysis
Data were entered into Microsoft Excel 2019, verified for consistency, and analysed using Epi Info version 7.2.7.0 (CDC, Atlanta, Georgia). Categorical variables are reported as frequencies and proportions. Continuous variables are presented as means with standard deviations (SDs) or medians with interquartile ranges, depending on distributional normality assessed by the Shapiro-Wilk test. No inferential hypothesis testing was prespecified, as this was a descriptive pharmacovigilance study. Institutional Human Research Ethics Committee approval was obtained (reference number: [IHEC/DPSRU/2023/XX]), and all participants provided written informed consent prior to enrolment.
Results
Study Population and Baseline Characteristics
Of 143 screened patients, 102 met eligibility criteria and constitute the analytic cohort (Figure 1). The cohort comprised 52 men (51.0%) and 50 women (49.0%), with a mean age of 34.7 ± 14.3 years (range 18-74 years). The largest age group was 21 to 40 years (47.1%), followed by 41 to 60 years (24.5%), ≤20 years (19.6%), and >61 years (8.8%). Twenty-two patients (21.6%) reported current or former tobacco use. MDR-TB was confirmed in 87 patients (85.3%) and XDR-TB in 15 (14.7%). Sixty-eight patients (66.7%) had a documented history of prior TB treatment, establishing drug-acquired resistance, while 34 (33.3%) presented with primary drug-resistant TB. Baseline demographic and clinical characteristics are summarized in Table 1.
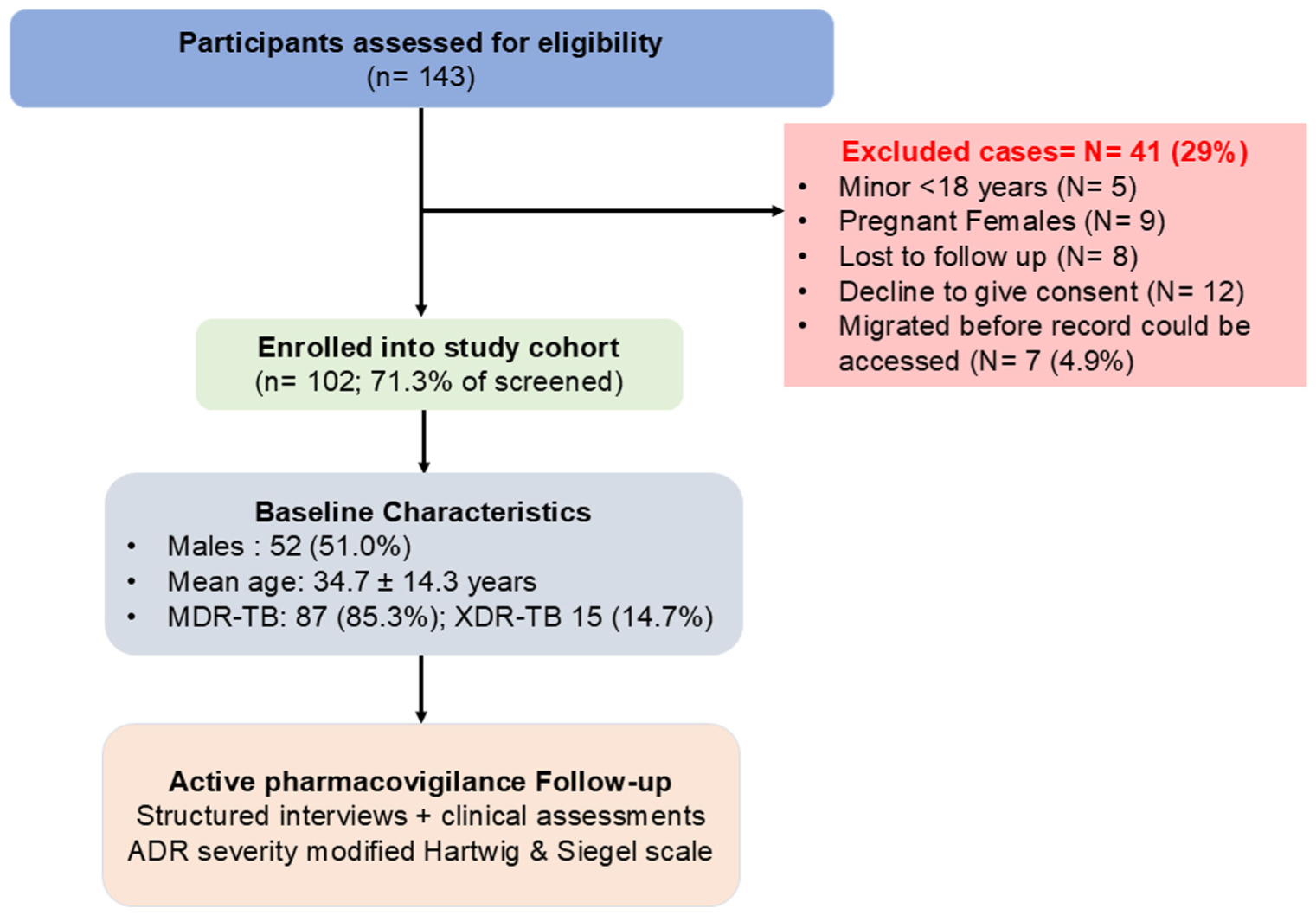
STROBE flow diagram of patient screening and enrolment for the study conducted between March 2023 and September 2023.
Baseline Demographic and Clinical Characteristics of the Study Cohort (N = 102).
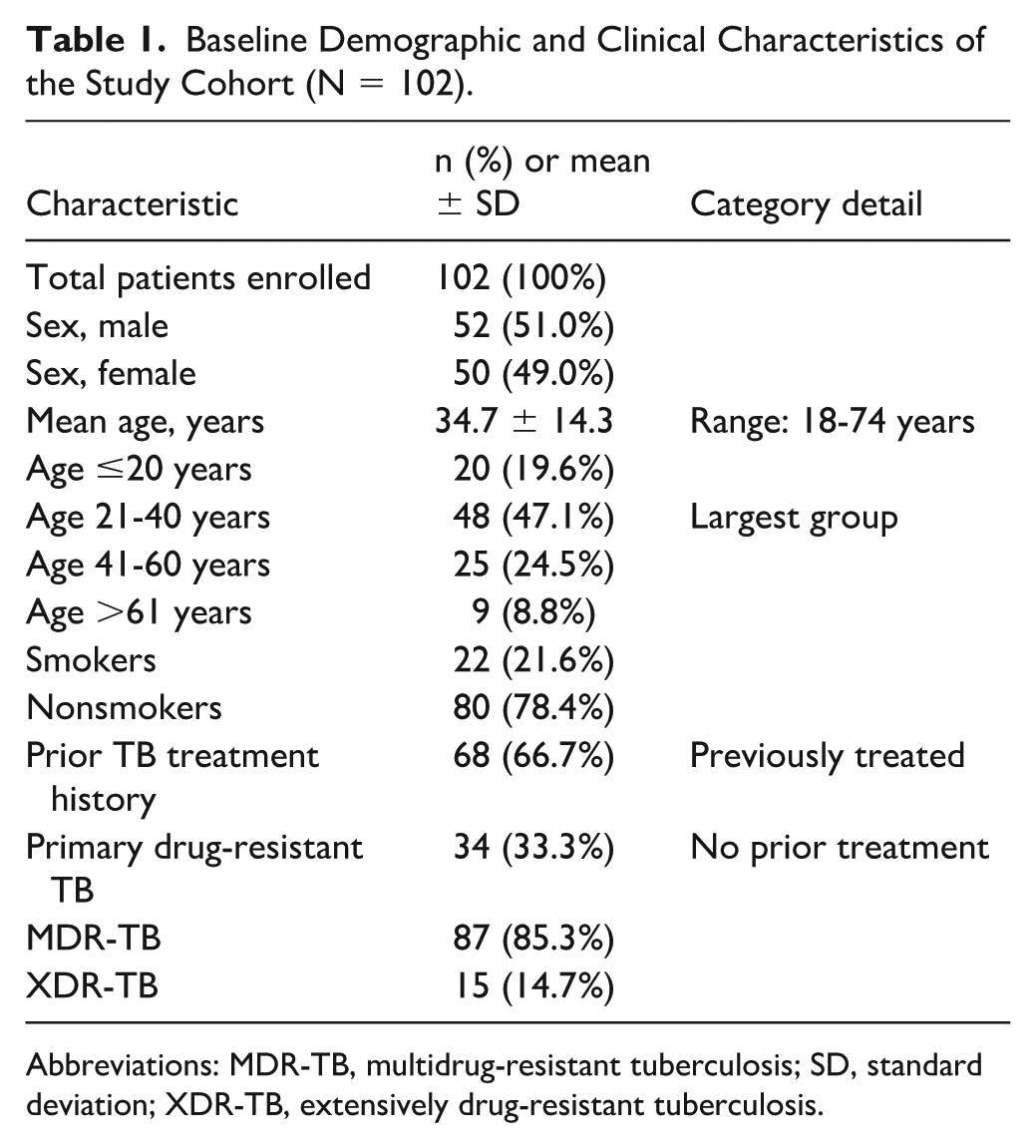
Abbreviations: MDR-TB, multidrug-resistant tuberculosis; SD, standard deviation; XDR-TB, extensively drug-resistant tuberculosis.
Treatment Outcomes
Treatment outcomes were available for all 102 enrolled patients. Favourable outcomes were achieved in 79 patients (77.5%), including 34 (43.0%) declared cured and 45 (57.0%) who completed treatment (Table 2). Unfavourable outcomes occurred in 23 patients (22.5%): 15 were lost to follow-up (65.2% of unfavourable outcomes), 7 experienced treatment failure (30.4%), and 1 demonstrated no clinical or bacteriological response by month 8 (4.3%). Importantly, no deaths occurred during the 20-month observation period. The overall treatment success rate of 77.5% compares favourably with the WHO-reported global MDR-TB benchmark of 56%. 1
Treatment Outcomes Among MDR/XDR-TB Patients Receiving Longer All-Oral Regimens (N = 102).
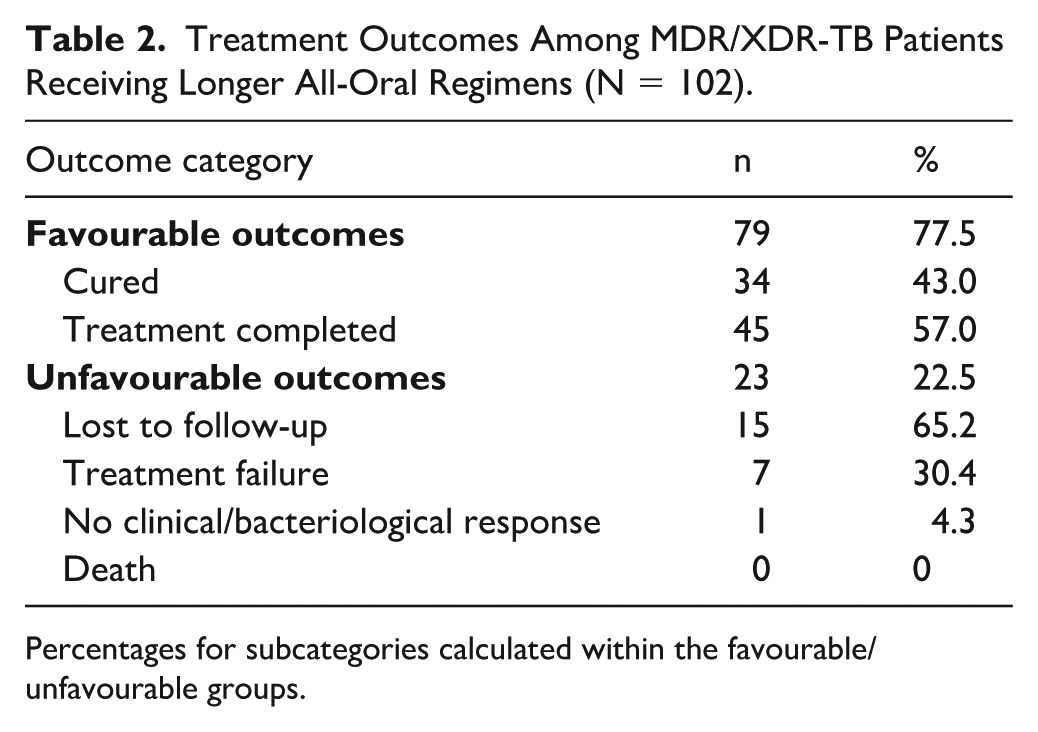
Percentages for subcategories calculated within the favourable/unfavourable groups.
ADR Severity Profile
A total of 158 ADRs were documented across 102 patients over the follow-up period. Grading by the modified Hartwig and Siegel scale classified 81% of ADRs as mild (levels 1-2; 95% confidence interval [CI] 74.2%-86.4%): 77 (48.7%; 95% CI 41.1%-56.5%) as level 1 (no treatment required) and 51 (32.3%; 95% CI 25.5%-39.9%) as level 2 (symptomatic management and no dose modification). Moderate ADRs accounted for 19.0% (95% CI 13.6%-25.8%): 22 (13.9%; 95% CI 9.4%-20.2%) at level 3 (requiring dose modification or temporary withdrawal) and 8 (5.1%; 95% CI 2.6%-9.7%) at level 4 (permanent drug discontinuation or hospitalization). No severe ADRs (levels 5-7) were recorded (Table 3). All CIs were calculated using the Wilson score method with proportions derived from the total number of ADRs (N = 158).
Distribution of ADR Severity According to the Modified Hartwig and Siegel Scale (N = 158 ADRs).

Abbreviation: ADR: adverse drug reaction.
ADR Distribution by System Organ Class
Adverse drug reactions affected 9 SOCs (Table 4). Gastrointestinal (GI) disorders were the most prevalent, documented in 57 events (36.1% of all ADRs; 95% CI 29.0%-43.8%; affecting 55.9% of patients): nausea or vomiting in 51 cases (89.5%) and abdominal discomfort in 6 cases (10.5%), predominantly attributable to levofloxacin and clofazimine. Of these 57 GI events, 49 (85.9%) were managed symptomatically without any regimen change—using antiemetics (ondansetron or metoclopramide), dietary advice (administering medications with food), and reassurance. The remaining 8 events (14.1%) required temporary dose reduction of the suspected offending agent (levofloxacin or clofazimine), with resolution in all cases within 2 to 4 weeks; none necessitated permanent drug discontinuation.
ADR Distribution by System Organ Class Using MedDRA Terminology (N = 102 Patients; 158 ADRs).

Percentages are within each system organ class.
Abbreviations: ADR, adverse drug reaction; Bdq, bedaquiline; Cfz, clofazimine; Cs, cycloserine; Lfx, levofloxacin; Lzd, linezolid.
Nervous system (NS) disorders (n = 46 events; 45.1% of patients) were the second most frequent category and of greatest clinical concern. Peripheral neuropathy attributable to linezolid was the most common moderate ADR (15 events; 32.6%; 95% CI 20.9%-47.0%), requiring dose reduction from 600 to 300 mg daily in most cases and permanent linezolid discontinuation in patients with progressive symptoms. Cycloserine contributed to nervousness (n = 8; 17.4%; 95% CI 9.1%-30.7%), restlessness (n = 5; 10.9%; 95% CI 4.7%-23.0%), and headache (n = 11; 23.9%; 95% CI 13.9%-37.9%), while levofloxacin accounted for dizziness (n = 7; 15.2%; 95% CI 7.6%-28.2%).
Skin and subcutaneous tissue disorders (n = 15; 14.7% of patients) were dominated by clofazimine-related hyperpigmentation in 13 cases (86.7%; 95% CI 62.1%-96.3%), which, while cosmetically distressing, was expected, dose-dependent, and partially reversible upon treatment completion. Skin rash was reported in 2 patients (13.3%; 95% CI 3.7%-37.9%), attributed to levofloxacin or linezolid.
General disorders (n = 12; 11.8% of patients) included fatigue or drowsiness (n = 9; 75.0%; 95% CI 46.8%-91.1%), asthenia (n = 2; 16.7%; 95% CI 4.7%-44.8%), and low-grade pyrexia (n = 1; 8.3%; 95% CI 1.5%-35.4%). Anorexia was the sole metabolic manifestation, occurring in 9 patients (8.8%; 95% CI 3.0%-10.5%), attributable to linezolid and clofazimine. Blood and lymphatic system disorders (n = 8; 5.1% of all ADRs; 95% CI 2.6%-9.7%) included linezolid-associated anaemia (n = 7; 87.5%) and pancytopenia (n = 1; 12.5%). Three patients developed severe anaemia requiring blood transfusion, while one patient experienced pancytopenia classified as a serious adverse event (grade 4 haematological toxicity; level 4 on the Hartwig severity scale). In all such cases, linezolid was temporarily suspended and subsequently reintroduced at a reduced dose following haematological recovery. Musculoskeletal events (n = 4) comprised arthralgia (n = 3) and myalgia (n = 1). Ocular events (n = 4) included visual disturbances (n = 3) and one case of linezolid-associated optic neuritis, which was confirmed ophthalmologically and prompted permanent linezolid withdrawal.
Cardiac disorders were the least frequent (n = 3; 2.9%). Two patients experienced palpitations attributable to bedaquiline or levofloxacin. One patient developed asymptomatic QT prolongation (QTcF >500 ms) during bedaquiline therapy, confirmed by serial ECG monitoring. This was managed through dose interval adjustment and enhanced cardiac surveillance; bedaquiline was not discontinued. No ventricular arrhythmias occurred.
Discussion
This prospective pharmacovigilance study provides systematically collected, real-world safety and effectiveness data for longer all-oral MDR/XDR-TB regimens implemented under NTEP in a high-burden tertiary care setting in North India. Three principal findings merit emphasis: (1) a treatment success rate of 77.5% that substantially exceeds the global MDR-TB benchmark; (2) an ADR profile that was overwhelmingly mild and manageable under active surveillance; and (3) linezolid-associated neurotoxicity and hematotoxicity as the dominant drivers of clinically significant, moderate-severity ADRs.
The demographic composition of our cohort—mean age 34.7 years with 47.1% of patients in the economically active 21- to 40-year stratum—mirrors findings from comparable Indian studies13,14 and reflects the well-documented predilection of MDR-TB for young adults, driven by ongoing community transmission in densely populated, high-incidence settings. The near-equal sex distribution observed here is notably distinct from the male predominance reported in most global literature. 15 This may reflect improved health care-seeking behaviour among women under NTEP’s active case-finding initiatives and reduced stigma associated with facility-based TB treatment.
A prior treatment history in 66.7% of our cohort corroborates the established association between inadequate or incomplete prior therapy and the emergence of acquired drug resistance in India. 16 Conversely, primary drug resistance was documented in 33.3%, highlighting ongoing transmission of resistant strains within the community—a finding with important public health implications for contact tracing and preventive therapy strategies. From a pharmacovigilance perspective, patients with primary MDR-TB represent a population exposed to second-line agents de novo, and their ADR trajectory may differ from those previously exposed to antitubercular regimens.
The treatment success rate of 77.5% observed in this study compares favourably with both the global MDR-TB benchmark of 56% 1 and outcomes reported from comparable all-oral cohorts in West Java, Indonesia (63%). 17 The absence of death in this cohort during 20 months of follow-up is a clinically meaningful finding, potentially attributable to structured clinical oversight, proactive ADR management, and the elimination of injectable-associated cardiotoxicity and ototoxicity—the latter being a major driver of mortality in historical injectable-based cohorts. Loss to follow-up (14.7%) remains the predominant unresolved challenge, consistent with published evidence on the psychosocial barriers to adherence in long-duration MDR-TB regimens. 18 Targeted psychosocial support, digital adherence monitoring, and community health worker engagement are evidence-based strategies warranting programmatic integration.
The overall ADR incidence in our cohort (158 ADRs across 102 patients; 1.55 ADRs per patient) was consistent with previously published prospective and retrospective data from similar high-burden settings.9,10 The predominance of mild GI reactions reflects the known tolerability profile of fluoroquinolones and clofazimine and suggests that symptomatic management without regimen modification is clinically appropriate in the majority of cases. The high prevalence of GI ADRs (55.9% of patients) has implications for adherence counselling and the timing of medication administration relative to meals.
Linezolid-associated peripheral neuropathy emerged as the most clinically significant moderate ADR, consistent with its established safety profile and pharmacokinetic basis. 19 Cumulative mitochondrial toxicity from linezolid’s inhibition of mitochondrial protein synthesis underlies the progressive nature of peripheral neuropathy and optic neuritis observed in this cohort. The strategy of preemptive dose reduction at 6 to 8 months—from 600 to 300 mg daily—as implemented in this NTEP programme appears clinically rational and is consistent with emerging evidence supporting dose optimization to preserve efficacy while limiting toxicity. 19 Baseline neurological examination, monthly symptom review, and periodic nerve conduction studies should be incorporated as standard monitoring elements in programmatic settings.
Haematological toxicity, including anaemia (n = 7) and pancytopenia (n = 1), was exclusively attributable to linezolid through its bone marrow suppressive mechanism. These findings underscore the necessity of monthly complete blood count monitoring throughout linezolid therapy, with prespecified thresholds for dose modification or temporary suspension.
The single case of bedaquiline-associated QT prolongation demonstrates the value of systematic, protocol-driven ECG surveillance during bedaquiline-containing regimens. Bedaquiline’s QT-prolonging effect is a recognized, dose-dependent pharmacological action mediated through human ether-a-go-go-related gene (hERG) potassium channel blockade. QTc prolongation exceeding 500 ms or a QTc increase of >60 ms from baseline warrants bedaquiline interruption and cardiology review. 20 The successful management of this case without treatment discontinuation in our cohort reinforces that structured monitoring—rather than drug avoidance—is the appropriate risk-mitigation strategy.
The complete absence of ototoxicity in this cohort is consistent with the expected safety advantage of fully oral, injectable-free regimens and represents a qualitative advance over previous aminoglycoside-based protocols, in which irreversible hearing loss affected a substantial proportion of MDR-TB patients.10,21 From a regulatory and policy perspective, this finding further supports the WHO-aligned programmatic transition to all-oral regimens.
Limitations
This study has several limitations that should be considered when interpreting the findings. First, the single-centre design and relatively small sample size (n = 102) may limit generalizability to other geographic or programmatic contexts. Second, the observational study design precludes causal inference regarding specific ADR-drug relationships, which were assessed on clinical and pharmacological grounds rather than formal causality scoring algorithms (eg, Naranjo scale). Third, absence of a comparator arm prevents direct efficacy or safety comparison with injectable-based or shorter MDR-TB regimens. Fourth, pharmacokinetic monitoring (eg, linezolid plasma concentrations) was not performed, which would have strengthened ADR attribution and dose-optimization analyses. Fifth, patient-reported outcomes were subject to recall bias in interview-based data collection. These limitations notwithstanding, the prospective design, active ADR surveillance, systematic severity grading, and real-world programmatic context constitute meaningful methodological strengths that enhance the clinical and regulatory relevance of these findings.
Conclusions and Relevance
This prospective pharmacovigilance cohort study demonstrates that longer all-oral MDR/XDR-TB regimens can be implemented safely and effectively under routine NTEP programmatic conditions, achieving a treatment success rate of 77.5% without mortality. The ADR profile was characterized by predominantly mild, predictable, and manageable reactions amenable to symptomatic treatment without regimen modification in the majority of patients. Linezolid-associated neurotoxicity and haematological toxicity represent the principal moderate-severity safety signals warranting proactive, protocol-driven clinical monitoring. Bedaquiline-associated QT prolongation, while infrequent, requires mandatory ECG surveillance infrastructure within all NTEP implementation sites.
From a clinical and public health perspective, these findings validate the real-world tolerability of injectable-free MDR/XDR-TB treatment and provide the pharmacovigilance evidence base necessary to inform regulatory oversight and programmatic refinement. Scaling structured pharmacovigilance—including standardized ADR documentation, laboratory monitoring schedules, and digital reporting pathways—is a prerequisite for the safe national implementation of innovative antitubercular regimens. Future multicentre prospective studies with pharmacokinetic monitoring and patient-centred outcome assessment are warranted to consolidate and extend these findings.
Footnotes
Ethical Considerations
The study protocol was approved by the Institutional Human Research Ethics Committee of Delhi Pharmaceutical Sciences and Research University (Reference: IHEC/DPSRU/2023/XX) and was conducted in compliance with the Declaration of Helsinki. All participants provided written informed consent before enrolment.
Author Contributions
AK and MG conceived and designed the study. AK, PN, and SC conducted patient enrolment, data collection, and ADR documentation. KA provided clinical oversight and patient management. RR and AK performed statistical analysis. AK drafted the manuscript. KA, RR, PN, SC, and MG critically revised the manuscript for intellectual content. All authors approved the final version for submission and accept accountability for all aspects of the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Deidentified data supporting the findings of this study are available from the corresponding author upon reasonable request and subject to institutional data governance approval.