
Abstract
Adrenocortical carcinomas are relatively rare, but they are considered to be highly aggressive malignant tumors. Sarcomatoid carcinomas represent an even more aggressive type. Bilateral malignant adrenal tumors are extraordinary rare, except for those that represent metastatic spread from a primary neoplasm. Here we report a case of a 69-year-old woman who presented symptoms that raised strong suspicions of adrenal insufficiency. Bilateral adrenal masses, identified in the imaging study, were responsible for the clinical manifestation and surgically resected. Surgical specimens of the bilateral adrenal tumors shared histological features compatible with sarcomatoid carcinoma. It was very difficult to confirm that the sarcomatoid carcinomas were derived from the cortex of the adrenal glands, but careful morphological observation and the panel of antibodies used for immunohistochemistry made the diagnosis possible. This is the first report of sarcomatoid carcinomas involving both adrenal glands. It should be emphasized that sarcomatoid carcinoma can arise bilaterally from even functionally impaired adrenal glands.
Introduction
Adrenocortical carcinoma is a high-grade and aggressive tumor. 1 It comprises only about 0.1 cases per 100 000. 2 Additionally, adrenocortical sarcomatoid carcinoma is a dedifferentiated variant and even more aggressive. Only 18 cases have been reported to date in the literature published in English.3-19 Furthermore, except for metastatic tumors and malignant lymphoma, only 2% to 6% of adrenal gland tumors are bilateral.20,21 In multiple endocrine neoplasia type 1 (MEN1), approximately 40% of patients have been found to have bilateral adrenocortical tumors, but only 1 was a carcinoma. 22 To the best of our knowledge, there has been no report of bilateral adrenocortical sarcomatoid carcinoma. Here we present a case of bilateral sarcomatoid carcinoma found with adrenocortical insufficiency, and histological features with immunohistochemical findings are discussed.
Case Presentation
A 69-year-old woman was admitted to our hospital with symptoms resembling adrenal insufficiency such as general malaise with hypotension. She had no particular past medical problems. She had no other conditions prior to these symptoms. Her family history was noncontributory. Adrenal cortex hypofunction was confirmed by high adrenocorticotropic hormone (ACTH, 1840 pg/mL) in the peripheral blood, secondary to low serum cortisol (1 μg/dL). Computed tomography (CT) of the abdomen revealed bilateral swelling of the adrenal glands and no calcification was found (Figure 1A). Magnetic resonance imaging (MRI) demonstrated heterogeneous density within bilateral adrenal lesions, which slightly high signal intensity on T2WI (T2-weighted image; Figure 1B) and low signal intensity on T1WI (T1-weighted image; Figure 1C). Bleeding was not detected. No tumor mass was observed in the kidney by CT or MRI. Compared with CT images obtained 2 years prior, rapid adrenal gland growth in the past 6 months was suspected, which might be responsible for the overt manifestation of the symptoms related to adrenal insufficiency such as general malaise. In addition, clinical conditions affecting adrenal function were not identified. Likewise, an imaging study, including positron emission tomography CT (PET-CT), showed neither primary site, including kidney, nor metastatic spread to the adrenal glands. Because adrenal carcinoma seems to be less likely to develop bilaterally in general, and a low potential for infiltrating propensity was found in the present case, lymphoma of low malignant potential was the most likely diagnosis in this case.
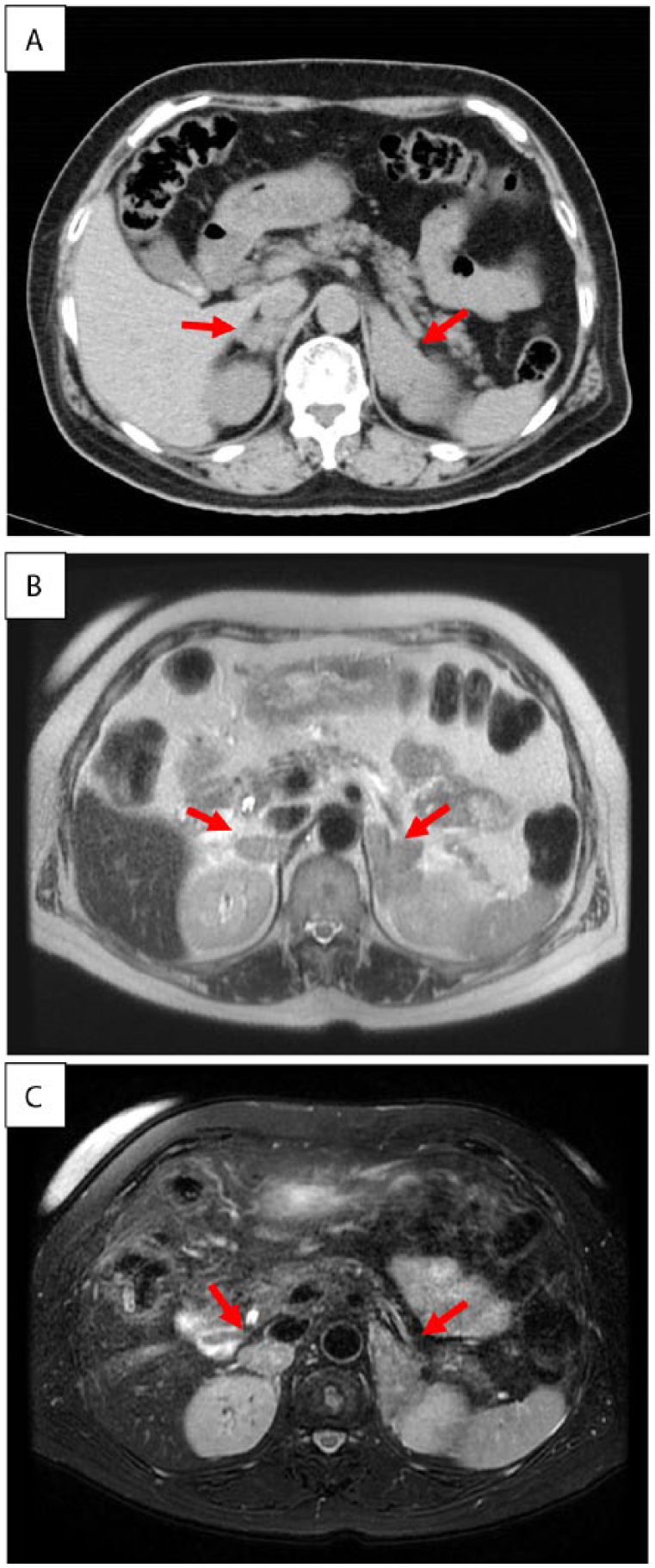
Computed tomography (CT) and magnetic resonance imaging (MRI) scan of abdomen. (A) Swelling of bilateral adrenal glands (arrows) can be seen in abdominal CT. (B) Interior of tumors (arrows) is heterogenic on T2-weighted image. (C) Low signal at T1-weighted image. Bleeding could not be detected in images.
After conventional steroid supplementation, bilateral adrenectomy was performed. Malignant lymphoma was easily excluded. The weights and sizes of the resected adrenal glands were 20 g, 5.5 × 3 × 3cm (right) and 35 g, 7 × 3.5 × 2 cm (left). Macroscopically, the cut surfaces of the adrenal glands showed white solid tumors that almost completely replaced the glands and invaded the surrounding soft tissue (Figure 2). The histological findings of the right adrenal tumor were exactly the same as those of the left tumor. Most of the proliferating cells were spindle-shaped and some had markedly pleomorphic or bizarre nuclei (Figure 3A). Storiform pattern (Figure 3B) of proliferation was also observed. There were some small areas with epithelioid arrangement that were only barely visible (Figure 3C) with transition to purely sarcomatous element (Figure 3D). Formalin-fixed, paraffin-embedded (FFPE) tissue was cut into sections of 3 μm thickness and immunostaining was performed with the primary antibodies according to the manufacturers’ instructions and visualized with a Ventana BenchMark ULTRA immunostainer (Ventana Medical Systems, Oro Valley, AZ, USA). Immunohistochemically, the tumor cells were diffusely positive for vimentin and CD99, and focally positive for S-100 (Figure 4A), cytokeratin CAM5.2 (Figure 4B), and calretinin (Figure 4C). The latter 2 antibodies were slightly positive in the area of epithelioid arrangement. Other types of cytokeratin AE1/AE3, CK7, and CD20 were all negative. RCC antigen and PAX8, which are renal cell markers, were both negative. Neuroendocrine markers: chromogranin A, synaptophsin, and CD56 were also negative. Other markers—TTF-1, GCDFP15, CDX2, SOX10, Melan A, HMB45, desmin, h-caldesmon, α-inhibin, α-smooth muscle actin, and SF-1 (Figure 4D) were all negative. The overall features were consistent with sarcomatoid carcinoma of the adrenal gland.
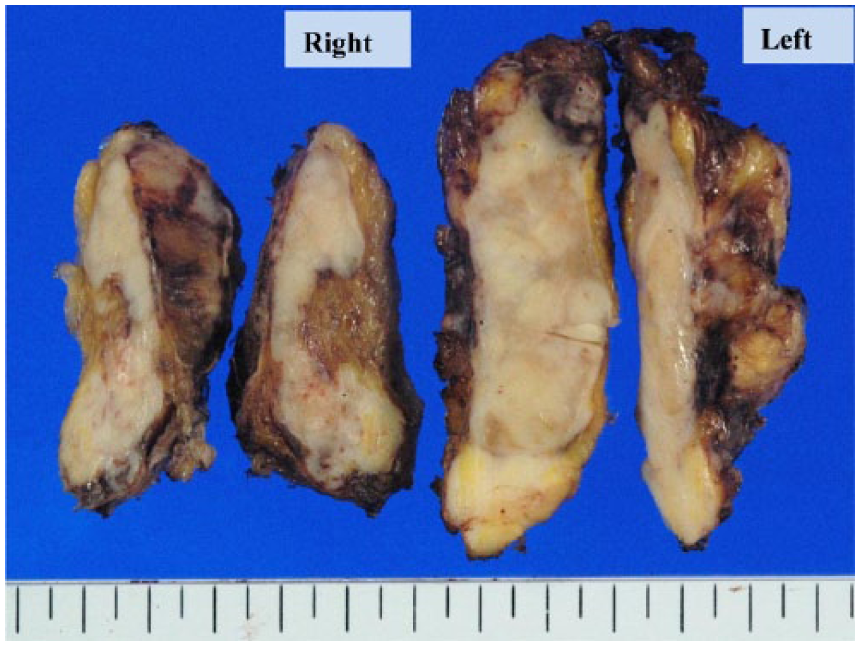
Macroscopic findings revealed massive proliferation with the tumors completely replacing the adrenal glands.
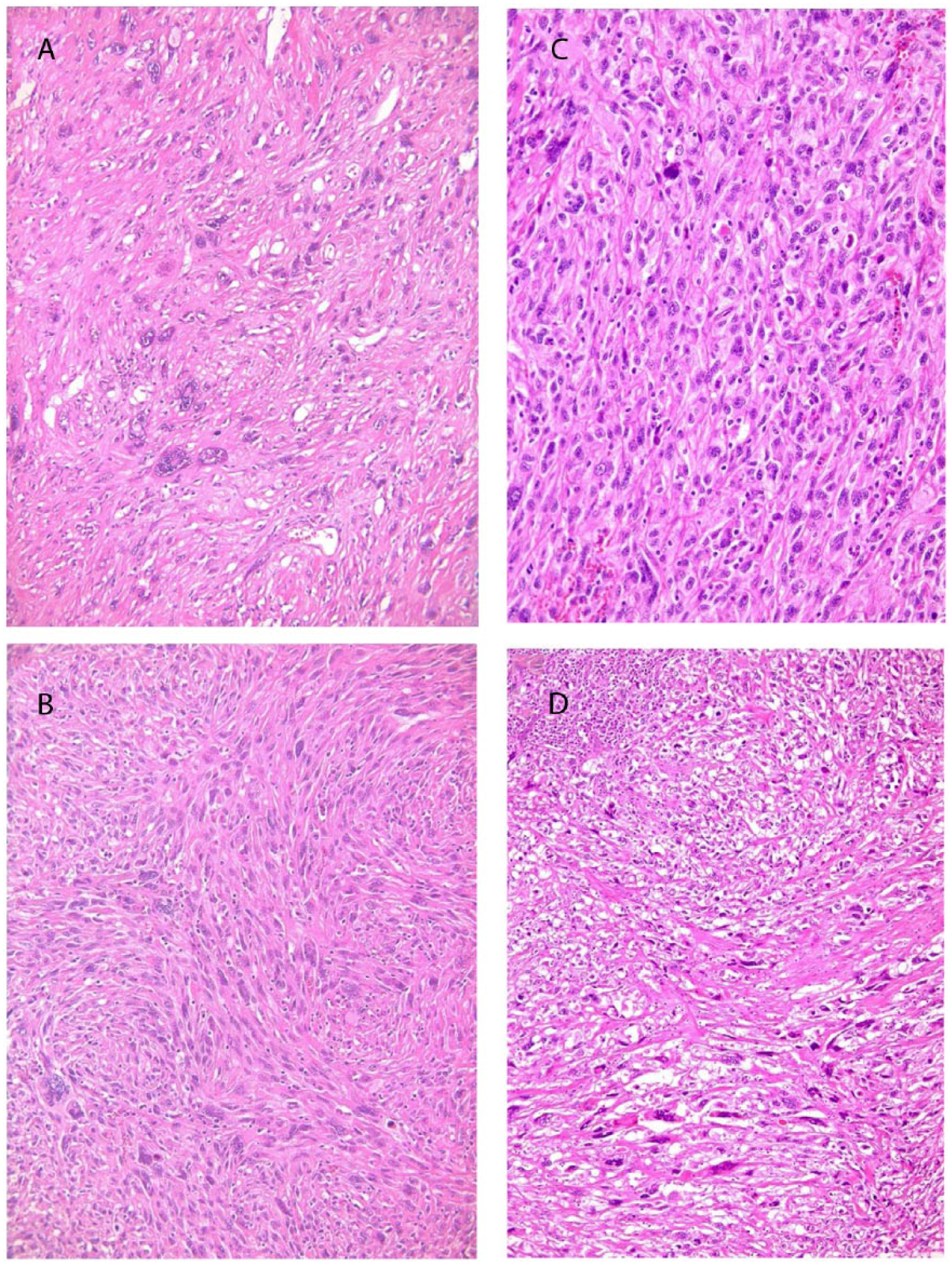
Microscopic findings of resected tumor. (A) Proliferation of tumors display spindle cells with frequently marked pleomorphic nuclei. (B) Display of storiform pattern. (C) A small area of epithelial-like arrangement is barely identifiable. (D) Transition between sarcoma and barely visible carcinoma elements.
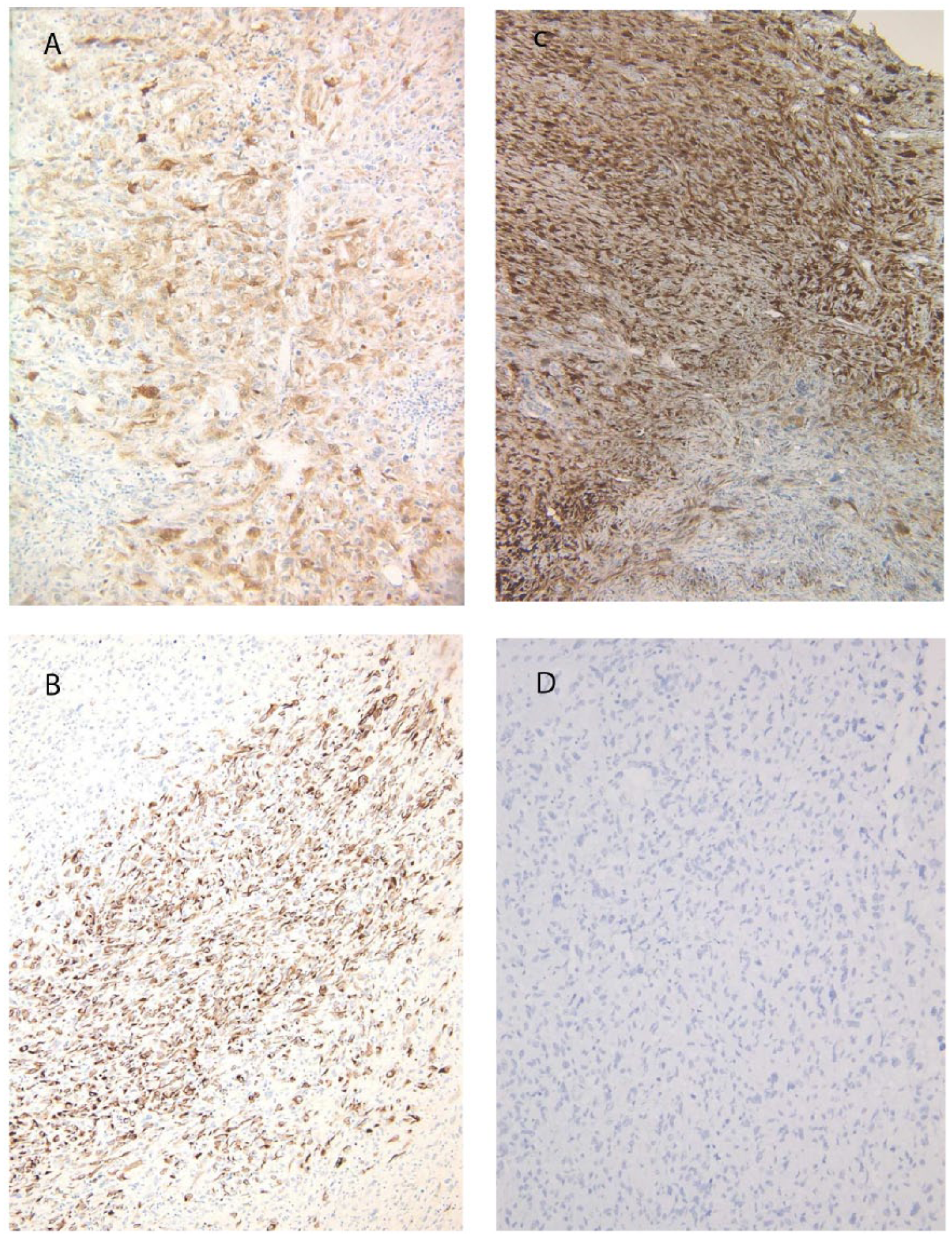
Immunostaining findings. (A) S-100, (B) CAM5.2, (C) calretinin, and (D) SF-1.
The patient’s general condition gradually deteriorated after the surgery, although steroid replacement therapy was continued. Three months later, the patient fell into septic shock caused by an abscess in the abdominal cavity. During drainage and antimicrobial treatment for the abscess, multiple metastatic tumors in the liver and abdominal lymph nodes were found by ultrasonography. One month after transfer to the palliative care ward, the patient’s respiratory condition became progressively worse and she died. Autopsy was not performed.
Discussion
According to the previous reports, some sarcomatoid carcinomas originating from the adrenal cortex contain various kinds of differentiated elements of sarcoma, such as osteosarcoma, chondrosarcoma and leiomyosarcoma, but others proliferate without any identifiable heterologous differentiation.17-19 It is not difficult to make a definitive diagnosis of sarcomatoid carcinoma if distinctive differentiation into adrenocortical cells is recognized. However, when such differentiation is indistinctive, the diagnosis can be very difficult, and immunostaining should be employed. Moreover, when tumors are present bilaterally, as seen in the present case, the situation becomes much more complicated. Calretinin, α-inhibin, synaptophysin, and melan A are known immunohistochemical markers of adrenocortical carcinoma, 23 and SF-1 has recently been reported to be an excellent marker.24,25 However, positive frequency for these markers varies depending on the antibodies used and the cases examined. It is often necessary to use a panel of various antibodies to specifically diagnose poorly differentiated tumors. As might be expected, the positive frequency of poorly differentiated carcinomas for cytokeratins is lower than well differentiated ones: The former are positive only faintly and focally, and sometimes negative for the antibodies. Fortunately, the bilateral tumors of the present case were obviously positive for CAM5.2, albeit focally. In the findings, epithelial differentiation was unclear initially on hematoxylin and eosin (HE)–stained sections. However, on careful review of the CAM5.2-positive areas, epithelial structures were recognizable even on HE-stained sectionsintermingled with high-grade sarcomatoid tumor cells, and the areas of transition from carcinoma to sarcomatoid tumor of no special type were also identifiable. Thus, when examining HE-stained sections, it is exceedingly important to search extensively for areas of epithelial differentiation before immunostaining for cytokeratins and adrenocortical markers. Positivity with calretinin and/or α-inhibin is known to be of highly diagnostic value, 23 although α-inhibin was negative in the present case. SF-1 has recently been reported to be excellent in its specificity for adrenocortical differentiation, compared to other antibodies, and it can also be a prognostic indicator.24,25 However, 2 of 8 cases of adrenocortical carcinoma, reported by Wanis and Kanthan, 18 were negative for SF-1 protein, and they were sarcomatoid carcinomas. Our case was also negative for SF-1, which might indicate expression of SF-1 protein decreases as the tumor undergoes dedifferentiation and be of little significance in differentiating sarcomatoid carcinoma from true sarcoma. In the present case, the tumor cells were positive for S-100 protein in the cytoplasm, indicating that the malignant peripheral nerve sheath tumor (MPNST), arising from adrenal gland, should be included in the differential diagnosis.26,27 However, even with partial differentiation into neurogenic cells confirmed by positivity for S-100 protein, sarcomatoid carcinoma can be distinguished from true MPNST, once again by careful morphological examination and a panel of antibodies including SOX-10, which has recently been demonstrated to be useful for diagnosis of MPNST. 28 The present case was negative for this protein.
Regarding bilaterality, metastasis was easily excluded from extra-adrenal organs in this case, but metastasis from one adrenal gland tumor to another could not be completely ruled out, although it was unlikely based on chronological images.
In conclusion, this is the first report of bilateral adrenocortical sarcomatoid carcinoma. We have to be aware that sarcomatoid carcinoma can arise in both adrenal glands synchronously, and in such cases, can present with the symptoms of adrenal insufficiency, whereas most adrenocortical carcinomas secrete steroid hormones. More data from similar cases need to be collected.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.