
Abstract
Intravascular large B-cell lymphoma (IVLBCL) is a subtype of diffuse large B-cell lymphoma, where the neoplastic lymphoid proliferation resides predominantly within the lumens of blood vessels but with no or few circulating neoplastic cells in the peripheral circulation. Focal or subtle involvement in some cases can cause the diagnosis to be misinterpreted or even overlooked, delaying the initiation of appropriate treatment. Our report focuses on a 78-year-old woman with a progressively enlarging thyroid mass, verified by ultrasound. She underwent a hemithyroidectomy, and microscopic evaluation demonstrated nodular thyroid parenchyma with atypical large cells in an intravascular distribution pattern identified on high magnification. Thorough evaluation showed that the large intravascular cells were positive CD20, PAX-5, and Ki-67 by immunoperoxidase staining, which lead to the diagnosis of IVLBCL. This case emphasizes the subtle appearance of IVLBCL, which may be missed on low-power light microscopy, and the need for careful evaluation of thyroid resection specimens.
Introduction
Intravascular large B-cell lymphoma (IVLBCL) is a distinct group of diffuse large B-cell lymphoma 1 where the neoplastic cells reside nearly exclusively within the lumens of small- and, less commonly, medium-size vessels. 2 First recognized in skin biopsies, 3 this entity may be seen throughout all organ systems with the central nervous system, skin, kidneys, lungs, and adrenal glands being most common. IVLBCL is known to occur within the thyroid; however, due to its infrequency, subtle morphologic findings, and variability of clinical symptoms, the disease can be missed or misdiagnosed, 4 subsequently delaying the appropriate treatment. These factors contribute to the overall dismal prognosis. As an example of this diagnostic dilemma, we present a case of IVLBCL in a 76-year-old woman presenting with a progressively enlarging thyroid nodule. Primary thyroid lymphoma is uncommon, accounting for less than 2% of primary thyroid malignancies, 5 and may have serious clinical consequences if undiagnosed.
Case Report
A 79-year-old woman presented with symptoms concerning for a transient ischemic attack. A neck ultrasound was performed to exclude carotid pathology, but revealed a circumscribed but sonographically concerning nodule in the left thyroid lobe, measuring 3.1 × 2.3 × 2.1 cm. The thyroid was nontender to palpation, and the patient did not have any obstructive symptoms. Neurological examination was normal. Her medical history was also significant for diabetes mellitus type 2, essential hypertension, and a history of an ovarian cyst.
A fine needle aspiration of the nodule was performed and interpreted as atypical epithelial cells of undetermined significance. She ultimately underwent a partial thyroidectomy to exclude a malignant neoplasm. Histologic examination revealed an atypical nodule with areas of solid and follicular architecture, cystic degeneration, and fresh hemorrhage (Figure 1). On high-power evaluation, follicular variant of papillary thyroid carcinoma could not be excluded due to mild nuclear enlargement and nuclear grooves. No nuclear pseudoinclusions were identified. Subtle clusters of large atypical cells were seen in a vaguely intravascular distribution pattern—apparent only on high-power magnification (Figure 2). Immunohistochemical staining demonstrated that these large neoplastic cells had a B-cell expression profile and were positive for CD45, CD20, PAX-5, p53, BCL6, and MUM-1 (Figure 3). Essentially, all of these cells were positive for Ki-67, indicating a very high proliferation index. CD5, CD10, CD30, BCL2, and Epstein-Barr virus in situ hybridization were negative.
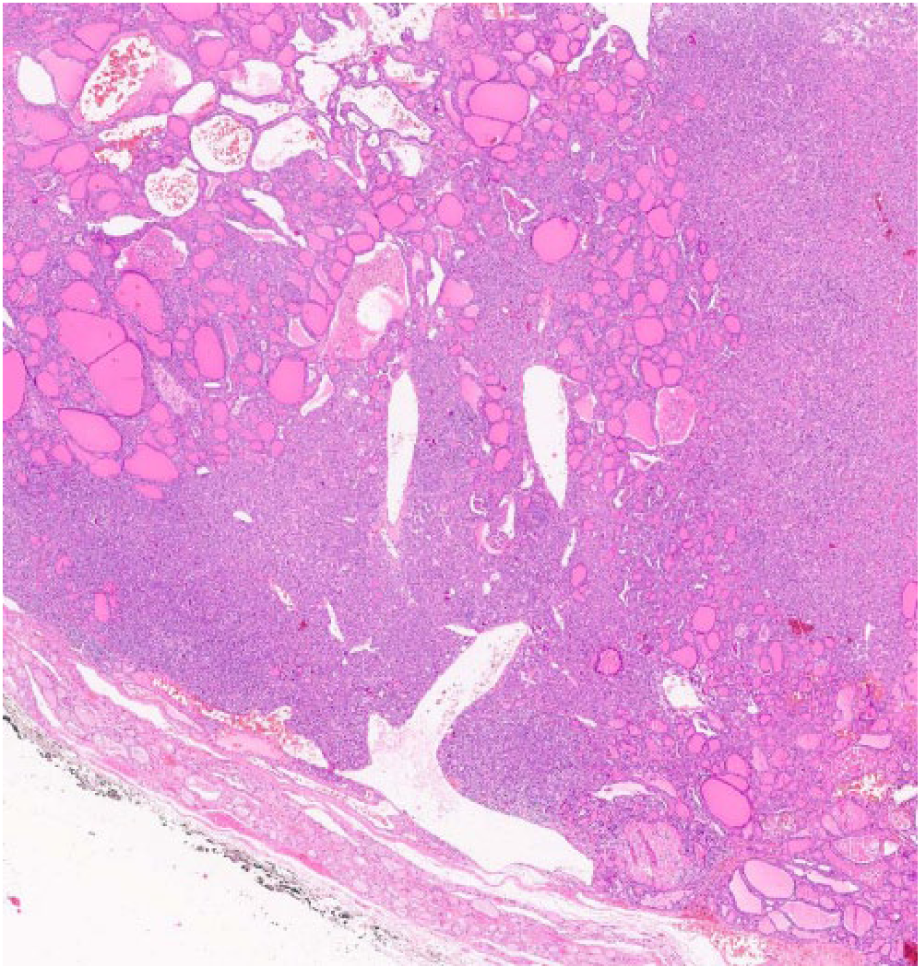
Low-power view of the atypical nodule with solid cellular component with interspersed follicles and cystic spaces and surrounding thin rim of residual normal thyroid tissue (hematoxylin-eosin; 2×)
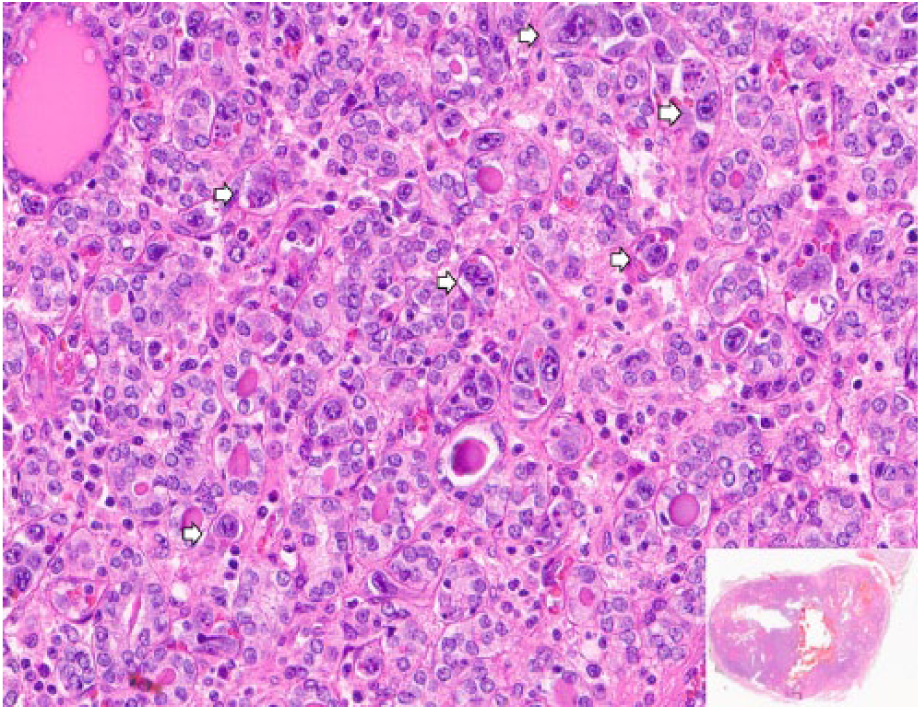
Histopathological features showing large cells with prominent nucleoli and irregular nuclear membranes (arrows), expanding small vasculatures (hematoxylin-eosin; 20×). A whole slide image is shown in the insert.
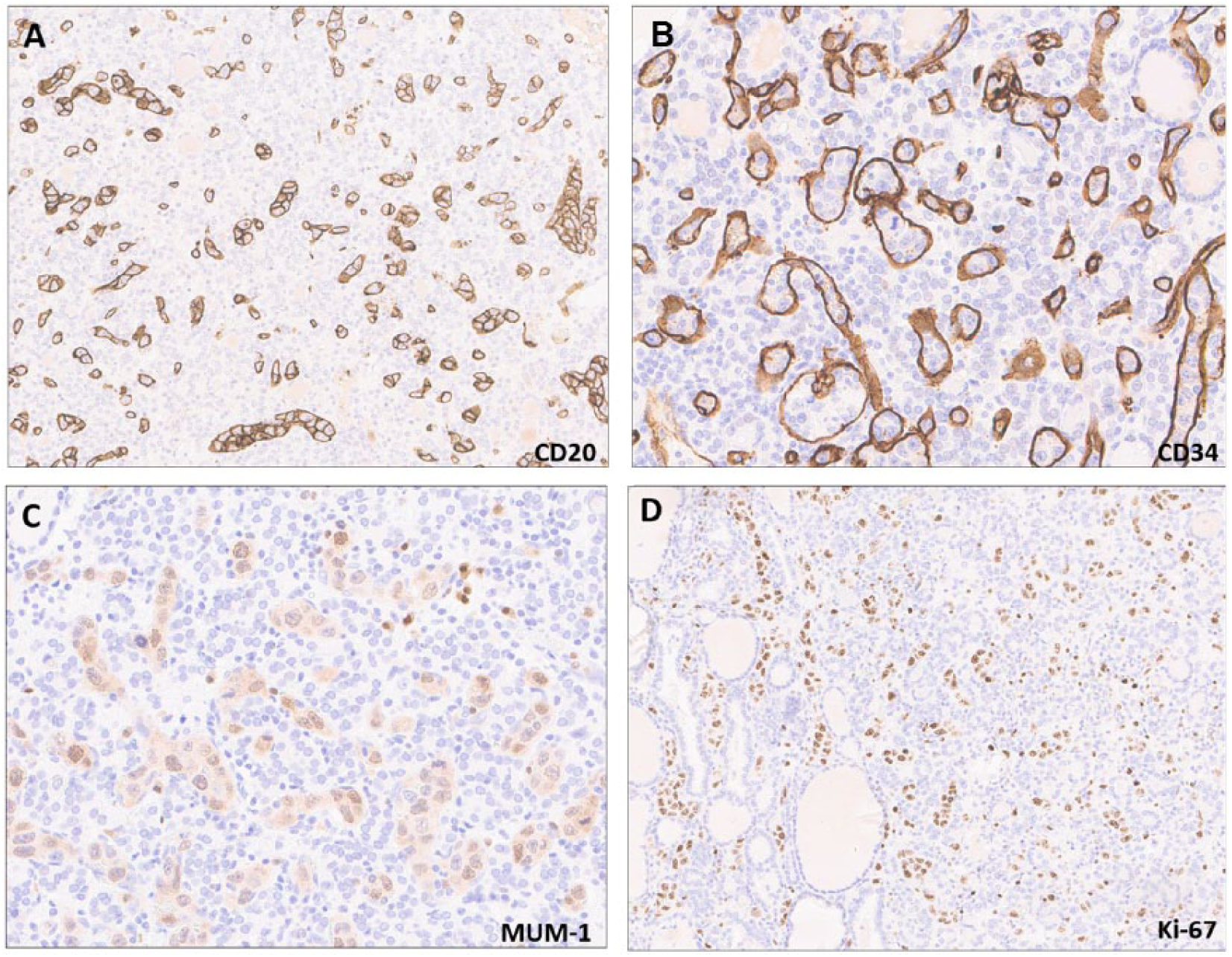
Immunohistochemistry revealed lesional cells to be of B-cell origin, positive for CD20 (A; 20×) and MUM-1 (C), located exclusively within small vessels, highlighted by CD34 (B; 20×). Immunoreactivity for Ki-67 in most nuclei (D; 10×).
The subsequent staging bone marrow biopsy was normocellular, showed normal hematopoiesis, and had no evidence of involvement by a lymphoproliferative neoplasm. She did not have peripheral cell count abnormalities, and serum lactate dehydrogenase levels were normal. A follow-up positron emission tomography-computed tomography showed mild hypermetabolic activity in the tumor resection bed, which was attributed to her postoperative state, and no other areas of hypermetabolic activity were identified. The patient underwent treatment with 3 cycles of R-CHOP with no evidence of disease recurrence after 6 years of close follow-up. This positive outcome is likely related to an early diagnosis and timely initiation of treatment.
Discussion
Lymphoma arising primarily in the thyroid is rare, accounting for 2% to 5% of all thyroid malignancies. 6 The most common subtypes of lymphomas arising in the thyroid gland are diffuse large B-cell lymphoma (50%), marginal zone lymphoma (10% to 23%), and follicular lymphoma (10%).7-10 About 60% to 90% of thyroid marginal zone lymphomas are associated with autoimmune lymphocytic (Hashimoto) thyroiditis, and the risk of transformation to lymphoma is estimated around 0.5%.4,10 T-cell lymphomas have been described as well but are exceedingly rare. 7 Overall, the prognosis for primary thyroid lymphomas is similar to that of comparable lymphomas arising in other anatomic sites.4,6,9
While a few cases of IVLBCL involving the thyroid have been reported,11-13 their presentation may be quite subtle making the diagnosis challenging for pathologists in general practice, and may be missed even by experienced physicians. Hematologic neoplasms are typically not a consideration when the decision to pursue a tissue biopsy is made.14-16
Patients typically present in the sixth or seventh decade. Depending on the clinical presentation, IVLBCL is conventionally subclassified into classical and “hemophagocytic syndrome-associated” variants. This latter grouping is characterized by an unfavorable prognosis and aggressive clinical course, and is more frequently described in patients of Asian descent. In addition to hemophagocytosis, patients often have fever, anemia, thrombocytopenia, hepatosplenomegaly, bone marrow involvement, and disseminated intravascular coagulation. However, the hemophagocytic syndrome-associated variant usually lacks skin lesions or neurological abnormalities.17-19 Chromosomal abnormalities such as 8q21, 19q13, and 18 trisomy have been reported in lymphomas with overwhelming systemic inflammatory syndromes. 20
While the neoplastic lymphoid proliferation occurs primarily within the lumens of small vessels, rare cases with large vessel involvement have been reported.2,21 The intraluminal distribution is attributed to CXCR3 expression on the malignant lymphomatous cells. CXCR3 has a high affinity for CXCL9, which is expressed on endothelial cells. 22 Lack of ICAM-1, β1-integrin, LFA-1 (lymphocyte function-associated antigen-1), or other molecules related to transvascular migration and lymphocyte homing have been reported as well.23,24 The rich vascular supply of the thyroid gland and adenomatous nodule microenvironment may induce overexpression of these chemokines on endothelial cells locally, leading to early preclinical thyroid gland involvement and incidental diagnosis in asymptomatic patients. 11 The t(3;14)(q27;q32) translocation, resulting in IGH/BCL6 fusion product, might play a role in IVLBCL lymphomagenesis. 25
The characteristics of this neoplasm generally preclude accurate diagnosis on fine needle aspiration biopsy, as demonstrated in our case. Although the diagnosis of IVLBCL is typically not on the differential diagnosis, a hemithyroidectomy or total thyroidectomy is usually necessary to correctly identify the disease process, especially for presumed stage I or infective endocarditis disease.9,26
In summary, IVLBCL is a rare and aggressive mature B-cell neoplasm with a nonspecific clinical presentation that is challenging to diagnose. The correct antemortem diagnosis is often missed, and the lymphoma is only identified at autopsy in about 16% of cases. 15 The 2-year survival rate of this rare entity has been reported to be 66%. Twenty-seven percent of patients reach the 3-year survival mark, and the median overall survival is 13 to 18 months.16,27 Early diagnosis of IVLBCL can potentially lead to better treatment response and outcomes, especially with the addition of rituximab and other targeted pharmacologic agents to the standard practice. 16 Considering this entity in the differential of a subtle atypical thyroid nodule could mark the difference between a timely diagnosis and a potential pitfall.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.