
Abstract
Mechanistic target of rapamycin kinase (mTOR) is a member of the phosphatidylinositol-3-hydroxide kinase (PI3 K)-related protein kinase family that functions as a central regulator of cell growth, metabolism, proliferation, and survival. The role of the TSC-mTOR signaling pathway in kidney tumors has been implicated in some hamartoma syndromes; however, with the advent and wide utilization of molecular studies, a growing number of kidney tumors have been linked to somatic or germline mutations involving genes that encode for this pathway, including eosinophilic solid and cystic renal cell carcinoma, low-grade oncocytic tumor, eosinophilic vacuolated tumor, renal cell carcinoma with fibromyomatous stroma and angiomyolipoma, among others. Herein, we review the contemporary developments of mTOR pathway-related renal neoplasia, focusing on the clinicopathologic features of the tumor entities.
Introduction
Mechanistic target of rapamycin kinase (mTOR), also referred to as the mammalian target of rapamycin, is a serine/threonine protein kinase, which is a member of the phosphatidylinositol-3-hydroxide kinase (PI3 K)-related protein kinase family. It is a key component of two distinct complexes, mTOR complex 1 (mTORC1) and complex 2 (mTORC2, which functions as a central regulator of cell growth, metabolism, proliferation, and survival). The mTORC1 is indirectly regulated by TSC complexes 1 and 2, which exert their inhibitory effect through Ras-related GTPase Rheb (Ras homolog enriched in the brain). Many pathways converge on TSC complexes and therefore affecting the mTOR function. These include growth factors, WNT signaling, hypoxia, and AMP-activated protein kinase.1–5 In addition to the TSC-Rheb-mediated regulation, mTORC1 can be directly activated by specific amino acids (Figure 1).6,7
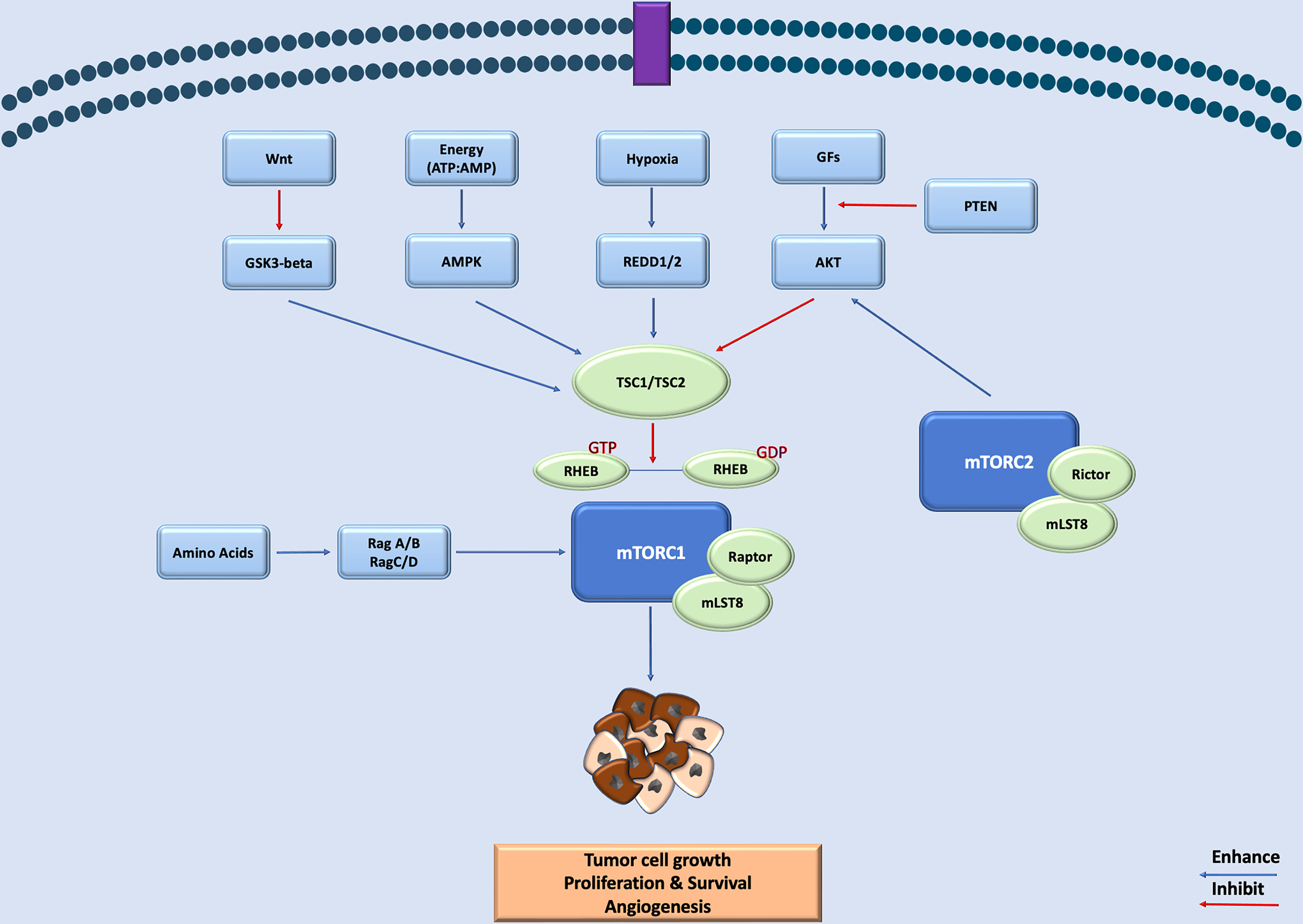
A schematic representation of the MTOR signaling pathway in tumor development and survival. mTOR, mechanistic target of rapamycin kinase.
The role of the TSC-mTOR signaling pathway in kidney tumors has long been implicated in some hamartoma syndromes, specifically tuberous sclerosis. In recent years, a growing number of kidney tumors, including carcinomas, have recently been linked to somatic or germline mutations involving genes that encode for this pathway's components, including eosinophilic solid and cystic renal cell carcinoma, low-grade oncocytic tumor, eosinophilic vacuolated tumor (EVT), renal cell carcinoma (RCC) with fibromyomatous stroma and angiomyolipoma (AML) (Table 1). Acquired cystic disease (ACD) associated-RCC, xanthomatous giant cell RCC, and sclerosing TSC1 mutated RCC have also been reported to have mTOR pathway alterations. It, however, remains unclear if the latter 2 represent standalone entities or extreme examples of EVT, TFEB-altered RCC, epithelioid AML, or more likely ESC-RCC.8–10
A Summary of the Histomorphologic and Immunohistochemical Features of mTOR Pathway Altered Renal Tumors.
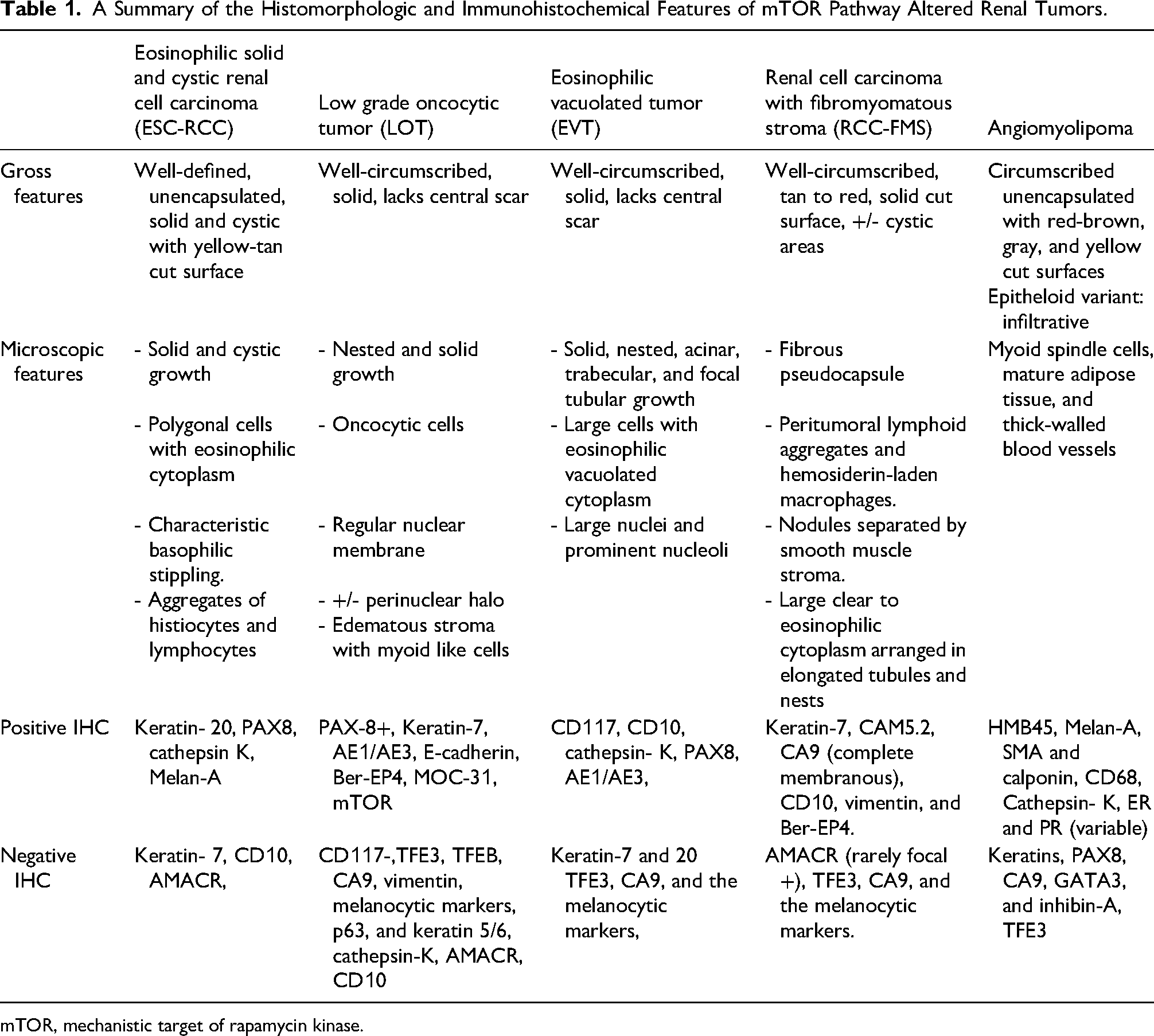
mTOR, mechanistic target of rapamycin kinase.
Herein, we review the contemporary developments of mTOR pathway-related renal neoplasia, focusing on the clinicopathologic features of the tumor entities.
Eosinophilic Solid and Cystic Renal Cell Carcinoma
Eosinophilic solid and cystic renal cell carcinoma (ESC-RCC) has been recognized as a distinct entity in the 2022 WHO classification of the urinary and male genital system. 11 It was first described as granular eosinophilic-macrocystic RCC in middle-aged to older women (median age, 55 years) patients with tuberous sclerosis. 12 Later, sporadic and several pediatric tumors were identified.13–15
On gross examination, most ESC-RCCs are well-defined, unencapsulated solid and cystic masses with a tan-brown or yellow cut surface, although tumors with a completely solid or dominantly cystic appearance were reported. 14 Multifocality was reported in nearly a third of tumors.14,16 Microscopically, tumors are distinguished by their solid and cystic growth. The solid areas are composed of nests or compact acini of polygonal cells with voluminous eosinophilic cytoplasm exhibiting characteristic basophilic stippling. These are sometimes admixed with eosinophilic-purple globules surrounded by a clear rim reminiscent of Leishmaniasis, representing aggregates of the rough endoplasmic reticulum. The nuclei are round to oval with inconspicuous nucleoli. Aggregates of histiocytes and lymphocytes are frequently encountered in these areas. The cysts vary in size and are lined by a single layer of hobnailed and multinucleated cells that exhibit cytoplasmic and nuclear features similar to those in the solid areas. Morphologic variations include cell vacuolization, focal tubular or papillary growth, scattered calcifications, and psammoma bodies. Melanin pigment was reported in one tumor.13,16–18
On immunohistochemistry, the most distinct feature of ESC-RCC is the positive reaction for keratin-20, although up to 12% of the tumors were reported negative. Other positive markers include PAX8, cathepsin K (focal or diffuse), and Melan-A (variable). Additionally, keratin-7 is usually negative or may show focal staining in most of the reported tumors and interestingly less than 20% of the tumors demonstrate focal positive immunolabeling for ER and PR (Figure 2).13,14,19

Eosinophilic solid and cystic renal cell carcinoma. A representative H&E photo showing cystic structures separated by thick fibrous septum occupied by cells with abundant oncocytic cytoplasm, containing pleomorphic nuclei. Characteristic cytoplasmic stipplings are present.
Several studies have reported copy number alterations, including gains of chromosome 7 (7p21.2–7q36.2 in 42%-50% of tumors), chromosome 13 (13q14.2 in 33% of cases), chromosome 16 (16p13.3–16q23.1 in 33%-67% of tumors), and chromosome 19 (19p12 in 33% of tumors). Copy number losses of 22q11.23 (33%) and Xp11.21 (42%) were the most commonly reported. Interestingly, some of these chromosomes were also the most frequently involved by loss of heterozygosity; chromosomes 16p11.2–11.1 (75%), Xq11.1–13.1 (75%), Xq13.1–21.1 (33%), 11p11.2–11.11 (33%), 9q21.1–22.2 (33%), and 9q33.1 (33%).14,20
Genetically, ESC-RCC demonstrates a low mutational burden. 20 Studies showed biallelic loss of either the TSC1 or TSC2 genes in most or all tumors, along with mTOR activation evidenced by focal cathepsin-K and mTOR expression.19–23
The differential diagnosis of ESC-RCC includes TFEB-altered RCC, tubulocystic RCC, acquired cystic disease-associated RCC (ACD-RCC), fumarate hydratase (FH)- deficient RCC, papillary RCC (PRCC), and thyroid-like follicular renal cell carcinoma. TFEB-altered RCC shares immunomorphologic features with ESC-RCC, in which, both tumors have epithelioid eosinophilic cells with areas of solid and cystic morphology, in addition to the frequent keratin 20 positivity. Additionally, evidence suggests the presence of positive melanocytic markers (Melan-A and cathepsin K) in ESC-RCC, therefore, in controversial tumors, resorting to molecular testing would be helpful to reach the precise diagnosis, as in the series reported by Li et al, where 4 of the reclassified ESC-RCC were tested negative for TFEB alterations.16,18,24 Tubulocystic RCCs are discriminated from ESC-RCC by their microcystic areas and tightly cohesive tubules lined by eosinophilic cells with no cytoplasmic inclusions, and they are diffusely positive for keratin-7 by immunohistochemistry. ACD-RCC—seen in end-stage renal disease (ESRD)—is characterized by solid, microcystic, and macrocystic architecture; however, the tumor cells have prominent nucleoli along with abundant oxalate crystals. On immunohistochemistry, these tumors are positive for CD10 and AMACR and negative for keratin-20. 25 , 26 Thyroid-like follicular RCC is composed of thyroid follicular-like containing eosinophilic secretions lined by cells with moderate eosinophilic cytoplasm and is typically positive for keratin-7 and negative for keratin-20. They also have a different recurrent molecular alteration (EWSR1::PATZ1 fusion).
Although ESC-RCC typically exhibits an indolent clinical course, rare tumors with metastases to lymph nodes, bone, lung, and liver have been reported.15,16,18,27 A complete response to mTOR targeted therapy (Rapamycin analog Everolimus) has been described in a patient with metastatic TSC2 mutated tumor. 15
Low-Grade Oncocytic Tumor
Low-grade oncocytic tumor (LOT) is an emerging renal tumor distinguished by its oncocytic cells that exhibit low-grade nuclear morphology with the characteristic diffuse keratin-7 immunoreactivity and negative KIT immunohistochemical pattern. Trpkov et al. initially described 28 tumors that developed in sporadic setting; however, it was later described in the context of ESRD and TSC syndrome where multifocality was observed.28,29 This tumor appears to account for 0.1–0.4% of all renal tumors and nearly 4% of all oncocytic renal tumors.28,30–32
Grossly, LOT forms a well-circumscribed, unencapsulated, or partially encapsulated mass with mahogany or tan-colored solid cut surface that lacks central scarring.28,32 The microscopic appearance is characterized by compact nested and solid growth of oncocytic cells with centrally located, uniformly rounded to oval nuclei with occasional perinuclear halos. Nuclear membrane irregularities are generally lacking but may be focally encountered in rare instances.32–34 Areas of edematous stroma with cords or loose reticular patterns of cells with myoid cell-like morphology are also common. Other reported features include focal tubular, tubuloacinar, and trabecular growth. Importantly, LOT lacks papillary growth, coagulative tumor-type necrosis, multinucleation, mitotic figures, or significant nuclear pleomorphism.29,32
Immunohistochemically, LOT shows a uniform reactivity for PAX8, pankeratin with clone AE1/AE3, E-cadherin, Ber-EP4, MOC-31, and characteristically diffuse keratin-7 and GATA3 immunoreactivity.35–37 Reaction for KIT (CD117) is either negative or only focally positive (Figure 3). Other markers, such as TFE3, TFEB, CA9, vimentin, HMB45, Melan-A, p63, and keratin 5/6 are all negative while CD10 and AMACR can be focally positive although they are mostly negative.29,32 SDHB and FH show retained expression.29,31,32,38

Low-grade oncocytic tumor. (A–D) Representative H&E photos showing compact nested, solid, trabecular and microcystic growth of oncocytic cells with centrally located, uniformly rounded to oval nuclei with occasional (soft) perinuclear halos. Areas of edematous stroma are present. (E, F) Representative immunohistochemical photos showing positivity for keratin 7 (diffuse, strong) and negativity for KIT, respectively.
Copy number analysis showed no consistent chromosomal losses, deletions, or gains. Some studies identified deletions at 1p36.33, 19p13.3, and 19q13.11 in some tumors while others detected frequent gains at 6p, 7p, 7q, 18p, and 22q and loss of chromosome 1.28,29,33
However, genetic alterations of mTOR pathway genes with mTORC1 activation have been identified as characteristic findings in LOTs. Most mutations involving the MTOR gene are heterozygous with preservation of the wild-type allele. These findings are reflected by the positive mTOR IHC and negative cathepsin-K along with the overexpression of 4EBP1-P and S6KP and underexpression of FOCI1 reflecting mTOR activation.30,33
Mutations involving RHEB and TSC genes have been identified in less number of tumors and rare reports have described germline TSC1 mutations in patients with multiple LOTs but clinically silent TSC. Interestingly, LOT was found to represent up to 26.7% of oncocytic tumors reported in patients with TSC.31,35,39,30 Additionally, gene expression profiling showed that LOT clusters away from renal oncocytoma, eosinophilic chromophobe RCC (ChRCC), classic ChRCC, and normal kidney. 23
LOT should be differentiated from renal oncocytoma, eosinophilic ChRCC, SDH-deficient RCC, FH-deficient RCC, and MiTF family translocation RCC, Renal oncocytoma shares many histologic features with LOT; however, LOT lacks the central scar classically described in renal oncocytoma, and unlike oncocytoma, LOT cells demonstrate perinuclear halos. In addition, by immunohistochemistry, LOT shows a diffuse positive reaction for keratin-7 and lacks KIT immunoreactivity, whereas renal oncocytoma shows the opposite immunoreactivity pattern. The eosinophilic ChRCC is distinguished from LOT by its eosinophilic cells, wrinkled nuclei, and frequent nuclear binucleation, while LOT lacks nuclear wrinkling or only focally exhibits this feature. In addition, LOT is characterized by the hypocellular stroma, which is not seen in eosinophilic ChRCC. Immunohistochemistry for keratin-7 and CD117 easily solves the problem, where the ChRCC shows a diffuse reaction for KIT. SDH-deficient RCC features flocculent cytoplasm with vacuolation, lacks the perinuclear halos, and shows loss of SDH immunostaining along with negative KIT immunoreactivity. Other tumors to consider in the differential diagnosis include hybrid oncocytic tumors, and PRCC, especially the solid morphology, in which the tumor is positive for AMACR and vimentin, and importantly, negative for GATA3 by immunohistochemistry.
To date, all reported tumors behaved indolently, without evidence of recurrence or metastasis over a follow up range of 0 to 344 months. 40
Eosinophilic Vacuolated Tumor
EVT is a newly described entity that was included as an emerging entity in the WHO 2022 classification of renal neoplasia. 11 The tumor was first described as “high-grade oncocytic tumor” and as “sporadic RCC with eosinophilic and vacuolated cytoplasm”.41,42 Similar to LOT, the majority of EVTs have been identified in the general population, but rare tumors in patients with TSC have been reported as well.41–46 EVTs are typically asymptomatic, and most tumors are incidentally discovered during imaging. 42 It was reported more in females, usually aged 25 to 73 years.41,42 Grossly, EVTs appear as well-circumscribed, unencapsulated solid, gray, tan tumors that are typically small, but size as large as 11.5 cm has been reported.41–44
Morphologically, the tumors commonly show solid, compact nested, acinar or trabecular growth patterns with focal tubular, or tubulocystic architectures in a few tumors. The cells have voluminous eosinophilic cytoplasm, with peripheral condensation and prominent cell membranes resembling ChRCC. Importantly, the cells have large intracytoplasmic vacuoles, hence the term vacuolated, with round to oval nuclei with granular to coarse chromatin and prominent nucleoli (WHO/ISUP grade 3). Large inclusion-like nucleoli is another distinctive feature. Other morphologic findings such as hypocellular edematous stroma and multinucleated tumor cells may be present and a subset of tumors exhibit extensive/prominent calcification; however, mitotic figures, foamy histocytes, infiltrating lymphocytes, and necrosis are typically absent. 42
EVT has a distinctive immunohistochemical pattern, with most tumors having KIT and CD10 positivity, with the former's expression can be weak. Keratins-7 and 20 are negative, and when positive, they are only focal in rare cells, not exceeding 10%.41–44 EVT is also positive for antimitochondrial antigen-antibody, cathepsin-K, PAX8, and pankeratin (clone AE1/AE3), but is completely negative for vimentin, TFE3, CA9, and melanocytic markers (Figure 4).

Eosinophilic vacuolated tumor. (A–E) Representative H&E photos showing solid and compact nested of cells have voluminous clear to eosinophilic cytoplasm, containing intracytoplasmic vacuoles with pleomorphic nuclei and prominent nucleoli. (F) Representative immunohistochemical photo showing positivity for cathepsin K.
Molecular studies demonstrated non-overlapping mutations in MTOR, TSC2, and TSC1 associated with a low-mutational tumor burden. In one tumor, a RICTOR missense mutation coexisted with an MTOR mutation.43,44 Unlike LOT, EVT shows MTOR alteration that is accompanied by loss of chromosome 1, which carries the second copy of the MTOR gene. 31
The differential diagnosis of EVT is wide and includes those discussed with the LOT. Additionally, important differential diagnosis related to the morphologic and/ or immunohistologic features includes epithelioid AMLs, in which, the positivity for melanocytic markers, CD68, and variably smooth muscle markers along with negativity for PAX8 are helpful to make such distinction.
EVTs are associated with good prognosis with no disease recurrence or progression in the previously described tumors. 47
Renal Cell Carcinoma with Fibromyomatous Stroma
In the literature, renal cell carcinoma with fibromyomatous stroma (RCC-FMS) was described as renal angiomyoadenomatous tumor—like RCC, tuberous sclerosis—associated papillary-type RCC, and several other terms for renal tumors with prominent leiomyomatous stroma such as clear cell renal cell carcinoma (CCRCC) with smooth muscle stroma, RCC with clear cells, smooth muscle stroma and negativity for 3p deletion, renal adenomyomatous tumor, RCC with angioleiomyoma-like or leiomyomatous stroma
RCC-FMSs occur mostly in sporadic setting, over a wide age range, but predominante in adults in their fifth decade. Grossly, tumors are solitary, well-circumscribed, and have a tan to red, solid cut surface; however, some tumors may appear partially cystic with an average size of 2.5 cm.12,48,53,54
Microscopically, the tumors are typically surrounded by thick fibrous pseudocapsule with peritumoral lymphoid aggregates and hemosiderin-laden macrophages. They consist of nodules of elongated and branched tubules or nests of cells with voluminous clear to eosinophilic cytoplasm separated by a distinct mesenchymal smooth muscle stroma. Focal papillary architecture was reported in a subset of tumors. A characteristic morphology of a biphasic pattern of central tubules surrounded by collapsed acini was identified in more than a third of the tumors.
Immunohistochemically, tumors demonstrate diffuse keratin-7, CAM5.2 and complete CA9 membranous staining; however, focal prominent cup-shaped staining was reported in some tumors. 48 CD10 is at least moderately expressed. Additionally, keratin 20 is negative in most tumors; however, focal apical positivity in subset of tumor cells was reported.35,55 Cytoplasmic desmin reactivity is seen in the smooth muscle component in all tumors, and focal AMACR expression was seen in rare tumors (Figure 5).53,54 Molecularly, the majority of tumors exhibit mutations involving TSC1, TSC2, and MTOR.

Renal cell carcinoma with fibromyomatous stroma. (A, B) Representative H&E photos showing nodules of elongated and branched tubules and nests of cells with voluminous clear to eosinophilic cytoplasm separated by a distinct mesenchymal smooth muscle stroma. (C–E) Representative immunohistochemical photos showing positivity for keratin 7 (strong, diffuse), carbonic anhydrase 9 (strong, diffuse, cup-like), and high molecular weight keratin (moderate, diffuse), with desmin highlighting the fibromyomatous stroma (F).
Features of RCC-FMS can overlap with CCRCC and clear cell papillary renal cell tumor (CCPRCT). The collapsed acinar morphology at the periphery of the nodules in RCC-FMS and the diffuse keratin-7 positivity can differentiate RCC-FMS from CCRCC. On the other hand, CCRCC is characterized by nodules with prominent cell nests and vascular-rich network, typically lacks strong and diffuse keratin-7 immunoreactivity and has an underlying VHL inactivation mutation.
Both RCC-FMS and CCPRCT exhibit diffuse keratin-7 immunoreactivity, but unlike RCC-FMS, the cells in CCPRCT have scant cytoplasm with their nuclei polarized away from the basement membrane. The cup-shaped CA9 positivity is typical for CCPRCT, while CD10 is negative or rarely focally positive, contrasting with the RCC-FMS.56–59 Additionally, on molecular level, CCPRCT lacks TSC1, TSC2, MTOR genetic alterations.
ELOC- mutated renal cell carcinoma, a distinct entity recognized in the 2022 WHO classification of renal neoplasia also has overlapping morphologic and immunohistochemical features with RCC-FMS. 54 Therefore, distinguishing them can be impossible based on the morphologic and immunohistochemical characteristics and may require molecular testing for the specific genetic alteration, ELOC and/ or monosomy 8 in the former and TSC1, TSC2, or MTOR in the latter. 54
The vast majority of RCC-FMS had indolent behavior, however, metastasis to lymph nodes was reported, specifically in patients with TSC.35,60
Angiomyolipoma
AML is a nonepithelial tumor comprising approximately 1% of all resected renal tumors. Sporadic tumors constitute 80% of the tumors, while the remaining occur in the TSC setting, where they are often bilateral, larger than the sporadic counterparts, associated with renal cysts, combined with polycystic kidney disease (PKD) and diagnosed earlier in life, sometimes as early as childhood. 61
Grossly, classic tumors of AML present as circumscribed unencapsulated masses with red-brown, gray, and yellow cut surfaces; however, the epithelioid subtype is usually large and infiltrative. Morphologically, classic AML has three components in variable proportions: myoid spindle cells, mature adipose tissue, and thick-walled blood vessels without elastic lamina. The myoid cells are typically arranged around the blood vessels.
Several morphologic variations/subtypes were described: oncocytoma-like AMLs, an extremely rare subtype composed of sheets of polygonal cells with eosinophilic cytoplasm, 62 AMLs with epithelial cysts, a rare subtype of AML characterized by solid and cystic components with the cysts being lined by cuboidal or hobnail epithelial cells reminiscent of the renal tubular epithelium underlined by spindle cells and the solid areas consist of spindle leiomyomatous cells and thickened blood vessels,63–65 and lastly, epithelioid AMLs, another rare variation of AML consisting of at least 80% epithelioid cells. Epithelioid AML represents 4.6% of all resected AML, with the neoplastic cells arranged in nests separated by a rich vascular network. Areas of spindled morphology arranged in diffuse sheets can be seen.66–68
Immunohistochemistry shows characteristic coexpression of melanocytic (HMB45, Melan-A, and MITF) and smooth muscle markers (SMA and calponin) in the myoid and lipoid components. Cathepsin-K is consistently positive, and desmin, ER, and PR are variably expressed. Keratins, PAX8, CA9, GATA3, and inhibin-A are negative (Figure 6).65,69,70

Angiomyolipoma. (A–D) Representative H&E photos showing classic examples of angiomyolipoma containing the three components in variable proportions: myoid spindle cells, mature adipose tissue, and thick-walled blood vessels. (E) Representative H&E photo of angiomyolipoma with epithelial cyst characterized by cysts lined by cuboidal or hobnail epithelial cells reminiscent of renal tubular epithelium, positive for HMB45 by immunohistochemistry (F).
Molecularly, AML harbors inactivating mutations involving the TSC2 gene and less commonly TSC1.67,68,71 A subset of tumors are associated with PKD1/TSC2 contiguous gene deletion syndrome. 72 In a cohort of 5757 patients who underwent nephrectomy at Mayo Clinic, 231 (4%) had PKD, of which, renal neoplasia was found in 26 (11.3%) patients, which included AMLs (n = 8) and a wide spectrum of renal epithelium—derived neoplasms (n = 18). of the latter, some showed overlap with TSC-associated renal neoplasia such as RCC-FMS and LOT, while other tumors which has no overlap such as clear cell, papillary chromophobe or ACD—associated RCC. 73
Epithelioid AML can exhibit more aggressive behavior with frequent recurrences and metastases than other subtypes. While no morphologic features have been identified to predict the metastatic behavior, some molecular alterations have been identified and can serve as a potential predictors for behavior, including TP53, ATRX, RB1, APC, and NF1 alterations. Additionally, TFE3 amplification or arrangement has been reported in some epithelioid AML with more aggressive behavior.74–76
The most important differential diagnoses for AML include TFE3 or TFEB-rearranged renal cell carcinoma, given the overlapping immunohistochemical profile; however, PAX8 and CD68 (PGM1) are reliable tools for distinguishing between them. AML with leiomyomatous cells should be differentiated from leiomyoma and those with predominantly lipomatous components to be distinguished from other lipomatous tumors, including liposarcoma and when the malformed blood vessels predominate, vascular malformation enters the differential diagnosis as well.
AML with classic morphology follows a benign course, and the management depends on the size. Tumors with epithelioid and pleomorphic features may have a more aggressive course, especially when associated with the following histologic features: more than 70% atypical cells, tumor necrosis, two or more mitotic figures/10 high power fields, atypical mitotic figures, extrarenal extension, venous invasion, and tumor size more than 7 cm.77,78 Nonetheless, the behavior of epithelioid tumors somewhat varies across studies, with some showing rare aggressive behavior. 67
In summary, molecular alteration in the mTOR pathway has gained special interest in the recent years, indebted to the expansion of molecular testing of kidney tumors. Despite the shared molecular pathway, which may shed some light on follow-up and management, these tumors have different morphologic, immunohistochemical as well as clinical behavior supporting separating them into different tumor entities.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Review of literature.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.