
Abstract
The combination of ependymoma and gliosarcoma elements in the same tumor is extremely rare, and the molecular characteristics of these entities are not clear. Here, we present a rare aggressive brain tumor in a 12-year-old boy harboring a ZFTA::RELA gene fusion, a characteristic feature of supratentorial ependymomas. On the other hand, the histopathological, molecular, and methylation profiles were compatible with a diagnosis of a mesenchymal type, IDH wild-type glioblastoma multiforme (GBM). Additional somatic alterations provide evidence of RAS/MAPK signaling pathway activation. Overall, this report highlights the histopathological and molecular characteristics of a rare and aggressive glial tumor.
Keywords
Introduction
Gliosarcoma is a rare variant of IDH wild-type glioblastoma multiforme (GBM), an aggressive astrocytic tumor with a poor clinical outcome. 1 Histologically, it is characterized by both glial and sarcomatous elements, 2 while at the molecular level, it shares a spectrum of genomic variants with GBM.3–5 Ependymomas are rare glial tumors arising from ependymal cells and are classified according to molecular and anatomical features. ZFTA (formerly C11orf95) supratentorial ependymomas are a sub-group of ependymomas defined by the presence of ZFTA::RELA genomic translocation. The combination of ependymoma and gliosarcoma elements in the same tumor is extremely rare,6,7 and to date, no molecular evaluation of these tumors was done. Here, we present a rare ZFTA::RELA fusion gliosarcoma and its histopathological, molecular, and methylation profiles.
Case Report
A 12-year-old boy was referred to our institute from a peripheral hospital due to a recurrent brain tumor. One year prior to his admission, and after suffering from severe headache, vomiting, and abnormal gaze, he was diagnosed with a left parietal brain tumor (Figure 1A-C). The tumor was surgically resected, and the patient had received a 54 Gy irradiation and 6 course chemotherapy treatments. Nine months following diagnosis, the tumor locally recurred and the patient was referred to our institute for pathological consultation. A brain computed tomography (CT) and magnetic resonance imaging (MRI) (Figure 1D-F) revealed 2 contrast enhancing parietal masses with evidence of extra-cranial extension, suggesting disease recurrence. Consequently, the patient underwent additional tumor resection in our institute. Three weeks following the operation, MRI scans revealed another recurrent tumor mass adjacent to the parietal bone (not shown) and the patient underwent a third brain operation.
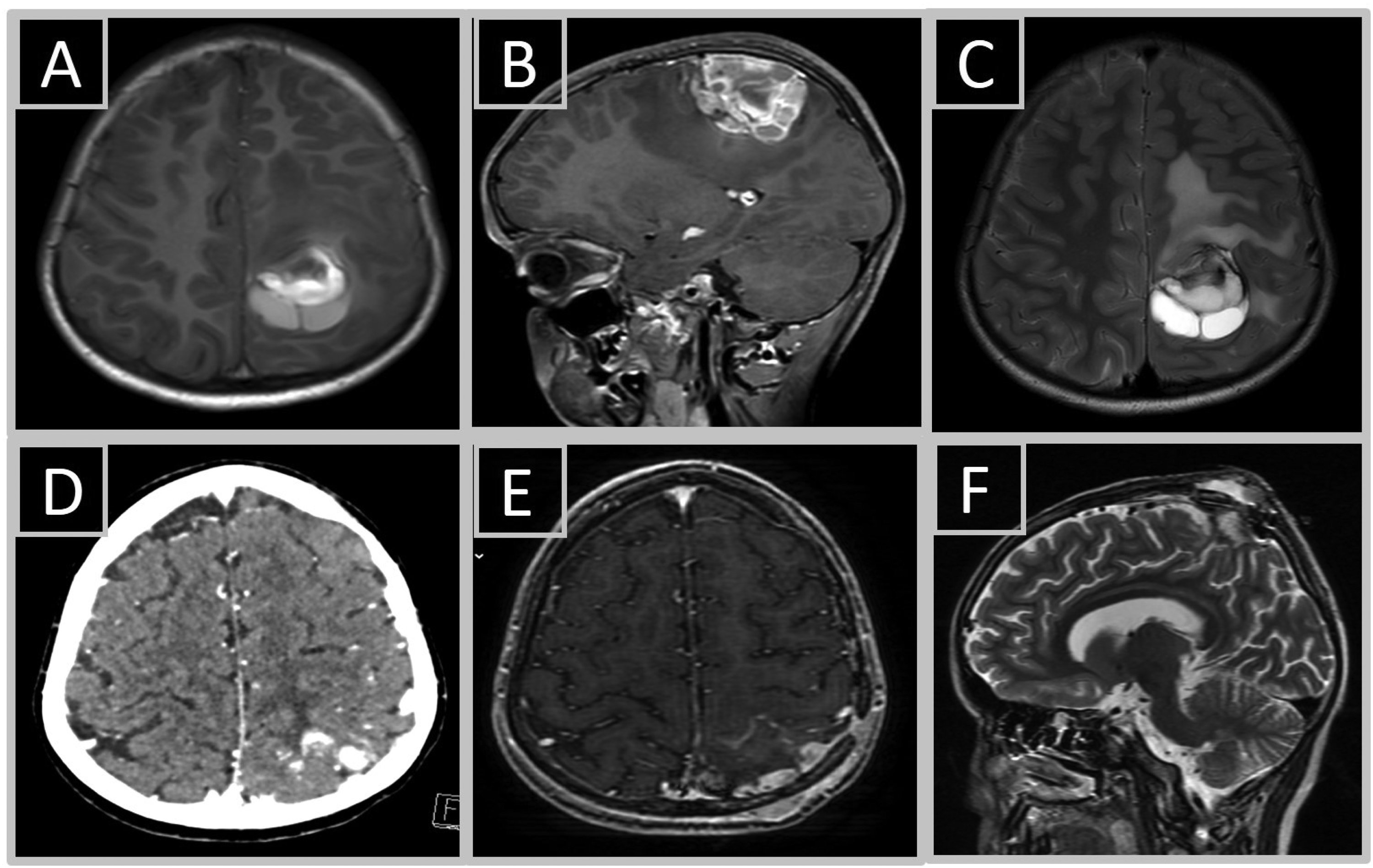
Tumors’ radiological characteristics. Brain MRI scans demonstrate a pre-operative T1-hyperintense lesion in axial (A) and sagittal (B) axes, and a T2-hyperintese lesion in axial axis (C). First disease recurrence as evident by the presence of 2 contrast enhancing parietal masses on CT scan (D), T1 axial (E), and T2 sagittal (F) MRI scans.
A revised histopathological examination of sections from the original tumor was performed. By histology, the original tumor was biphasic and composed of 2 cell types: spindle cells with marked atypia and 30 mitoses per 10 high power field (HPF), alongside another population of atypical epithelioid cells (Figure 2A-C). A few areas of necrosis surrounded by pseudopalisading cells were also observed. Of note, the epithelioid component was focally positive for glial fibrillary acidic protein (GFAP) immunostaining while the spindle cell component was negative (Figure 2D). Epithelial membrane antigen (EMA) immunostaining was focally positive. OLIG2, BCOR, and synaptophysin immunostaining were negative. Sections from the second operation (the first disease recurrence) demonstrated a pure spindle cell tumor (Figure 3A), which was negative for GFAP and EMA immunostaining. Next-generation sequencing (NGS) was performed on DNA and RNA extracted from sections of the second operation (the first disease recurrence). This was done using commercial NGS panels covering 501 genes and 97 genes in DNA and RNA libraries respectively (Thermo Fisher, A49667, A36486). A ZFTA::RELA fusion transcript and point mutations in HRAS, NF1, TP53, BCOR, and STAG2 genes were identified (Table 1). A targeted sequencing of the original tumor, the recurrent tumor, and a buccal swab germline control from the same patient was done. This analysis confirmed that all genomic alterations including ZFTA::RELA fusion were somatic (as they were not detected in the germline control sample) and were present in both the original and recurrent tumors. A strong nuclear staining for p65 was observed in tumor cells, confirming that this lesion is associated with ZFTA::RELA fusion (Figure 3B). Finally, DNA methylation profiles of the original and recurrent tumors were assessed using The Infinium Methylation EPIC Array (Illumina) and DKFZ Brain classifier version 12.8. A methylation signature of a glioblastoma IDH wildtype, mesenchymal subtype, was identified in both lesions (calibrated confidence score 0.95501). The final diagnosis of gliosarcoma was made. Unfortunately, a rapid disease progression eventually led to the patients’ death within 6 months from the pathological diagnosis.

Histopathological characteristics of the original tumor. Histological sections from the original tumor showing a biphasic lesion comprised of atypical epithelioid and spindle cells (A: 10×, B: 20×, C: 40×), focally immunoreactive for GFAP (D: 40×).
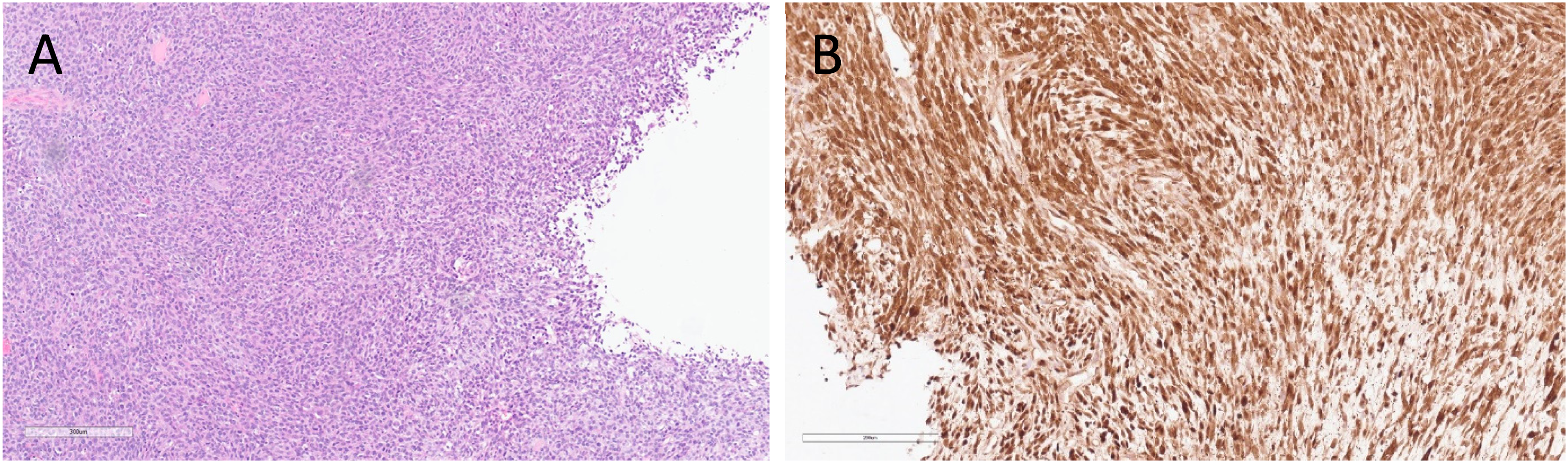
Recurrent tumor's histopathological and immunostaining characteristics. Histological sections from the first disease recurrence (the second operation) (A: 40×) showing spindle cell morphology. Tumor cells are strongly immunoreactive for Rela/p65 (B: 40×).
Mutations Identified by Next-generation Sequencing of Tumor's DNA.
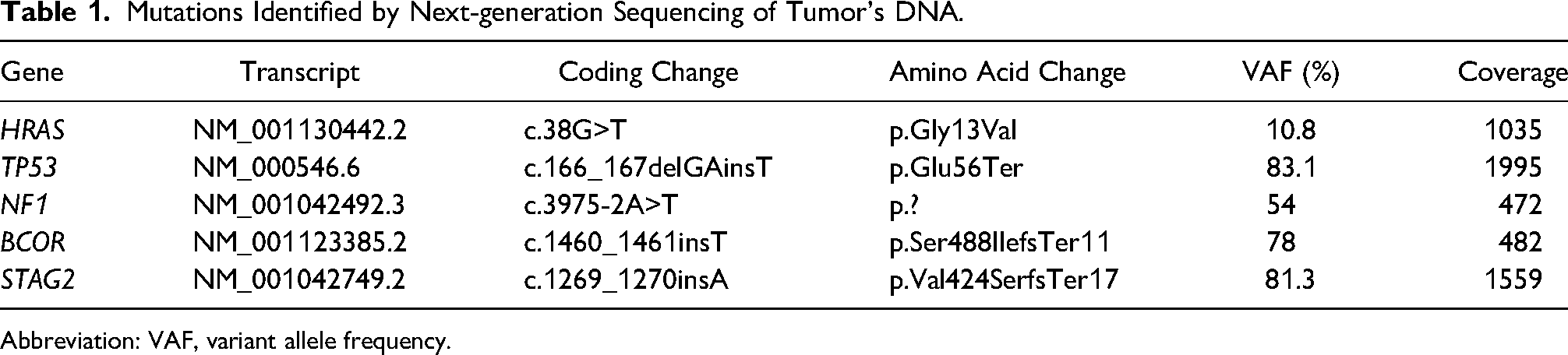
Abbreviation: VAF, variant allele frequency.
Discussion
The presented tumor exhibited histopathological and immunohistochemical features of a gliosarcoma, as it is a highly cellular and partially necrotic tumor containing both glial (GFAP-positive epithelioid cells) and sarcomatous (GFAP-negative spindle cells) elements. The classic histopathological and immunohistochemical characteristics of ependymoma (namely ependymal rosettes, perivascular pseudorosettes, or perinuclear dot-like EMA staining) were absent. Therefore, a diagnosis of ependymoma could not be made and the lesion was diagnosed as gliosarcoma. However, the presence of a ZFTA::RELA fusion in gliosarcoma or GBM is unusual, and to our knowledge was not previously described.
Rarely, ependymomas with sarcomatous differentiation have been reported,6–8 some of which harbored ZFTA::RELA fusion transcripts.6,9 Recently, a methylation and molecular profiling of a ZFTA::RELA fused ependymoma-like tumor with mesenchymal differentiation was described. 10 This and our reports provide similar evidence that ZFTA::RELA-driven mesenchymal brain lesions may not have ependymoma methylation profiles. However, in contrast to this previously described lesion, our tumor had a definitive methylation pattern of a mesenchymal subtype, IDH wild-type GBM, a diagnosis which is further supported by histopathological, immunohistochemical, and some molecular findings. For example, HRAS and NF1 somatic mutations are common genetic alterations in GBM3,11–15 and NF1 mutations are specifically enriched in mesenchymal-type GBMs. 16 On the other hand, common GBM alterations such as TERT promoter mutations, EGFR amplification, CDKN2A/B loss, or copy number changes in chromosomes 7 and 10 were absent in our tumor.
To conclude, in this report, we present a unique aggressive brain tumor in a 12-year-old patient characterized by histopathological and methylation profiles of a gliosarcoma combined with a ZFTA::RELA fusion. This is the first description of a gliosarcoma harboring ZFTA::RELA fusion and its broad molecular and methylation profiling. Importantly, some of the somatic mutations identified may shed light on key biological and evolutionary aspects of these tumors, such as aberrant activation of the RAS/RAF/MAPK signaling pathway. Consequently, the sensitivity of such rare tumors to MEK inhibition should be assessed and tested in future clinical trials.
Footnotes
Data Availability
All other relevant data are available from the corresponding author upon reasonable request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Ethics approval was given to this study by the Human Research Ethics Committee of the Rambam Health Care Campus under IRB number 0500-22-RMB-D.
Informed Consent
A waiver of informed consent was given by the Human Research Ethics committee of the Rambam Health Care Campus under IRB number 0500-22-RMB-D.
Trial Registration
Not applicable, because this article does not contain any clinical trials.