
Abstract
Melanin pigmentation in epithelioid angiomyolipoma (AML), a potentially malignant and locally aggressive renal tumor, is an exceptionally rare finding. Accurate differentiation from other pigmented tumors, such as melanoma, pigmented paraganglioma, and pigmented renal cell carcinoma is essential for proper diagnosis and treatment. A retrospective review of 2 departmental archives (2000-2023) identified 2 pigmented renal epithelioid AML specimens. Clinical presentations, histopathological features, immunohistochemical profiles, and molecular findings were analyzed. Both specimens exhibited melanin pigmentation, and 1 tumor demonstrated unique molecular alterations, including TSC2 and TP53 mutations. This study underscores the importance of meticulous histopathological evaluation and molecular profiling in diagnosing and managing this extremely rare variant of epithelioid AML.
Introduction
Epithelioid angiomyolipomas (AMLs) are rare renal tumors belonging to the perivascular epithelioid cell tumor (PEComa) family. While most AMLs are benign, epithelioid AMLs can exhibit locally aggressive and metastatic behavior, with metastasis reported in approximately 30% of tumors.1,2 These tumors have a strong predilection for women, with a 4:1 ratio of female-to-male patients, and are typically diagnosed in individuals in their 50s. 3 Epithelioid AMLs account for less than 1% of renal neoplasms,4,5 and an even rarer subset of these tumors contains melanin pigmentation.
To date, there have been 14 reported pigmented epithelioid AMLs of the kidney6–19 (Table 1). These tumors present significant diagnostic challenges due to their overlapping features with other pigmented tumors, such as melanoma and pigmented renal cell carcinoma. In this report, we present 2 additional pigmented epithelioid AMLs, providing detailed clinicopathological and molecular findings. Our aim is to improve diagnostic accuracy and management strategies for this exceptionally rare condition.
Pathological Features of Pigmented Renal Epithelioid Angiomyolipomas/Perivascular Epithelioid Cell Tumors.
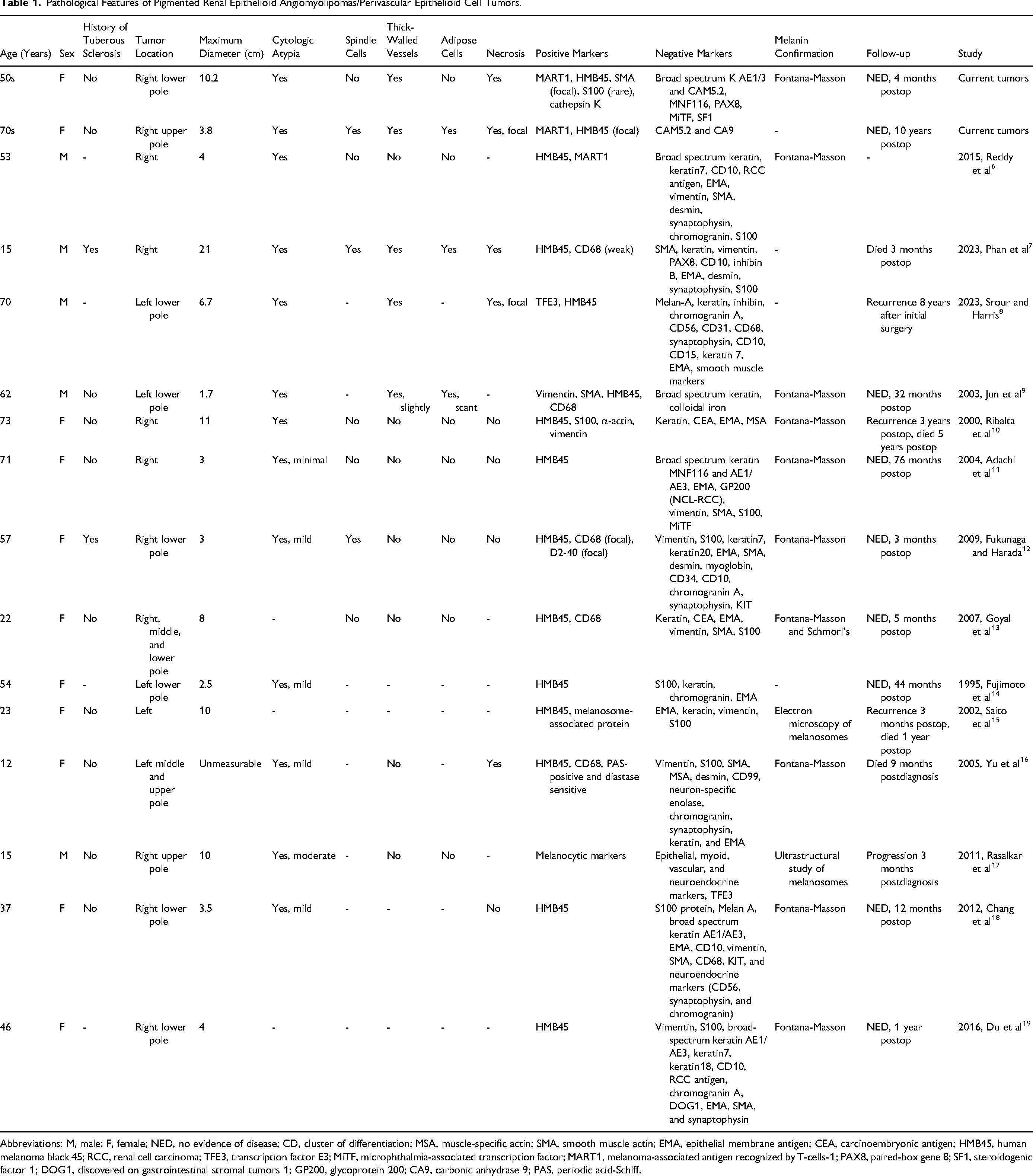
Abbreviations: M, male; F, female; NED, no evidence of disease; CD, cluster of differentiation; MSA, muscle-specific actin; SMA, smooth muscle actin; EMA, epithelial membrane antigen; CEA, carcinoembryonic antigen; HMB45, human melanoma black 45; RCC, renal cell carcinoma; TFE3, transcription factor E3; MiTF, microphthalmia-associated transcription factor; MART1, melanoma-associated antigen recognized by T-cells-1; PAX8, paired-box gene 8; SF1, steroidogenic factor 1; DOG1, discovered on gastrointestinal stromal tumors 1; GP200, glycoprotein 200; CA9, carbonic anhydrase 9; PAS, periodic acid-Schiff.
Materials and Methods
Specimen Identification
A retrospective review of 2 departmental archives (2000-2023) identified 2 pigmented renal epithelioid AMLs. Demographic and clinical data were retrieved from electronic medical records.
Immunohistochemical Analysis
Immunohistochemistry was performed on 4 μm sections from formalin-fixed, paraffin-embedded tissue using automated staining systems. A panel of melanocytic, smooth muscle, and renal markers was employed, including human melanoma black 45 (HMB45), melanoma-associated antigen recognized by T-cells-1 (MART1), microphthalmia-associated transcription factor (MiTF), S100, steroidogenic factor 1 (SF1), broad spectrum keratin, paired-box gene 8 (PAX8), carbonic anhydrase 9 (CA9), cluster of differentiation 68 (CD68), and smooth muscle actin (SMA). Positive and negative controls were included for quality assurance.
Molecular Profiling
Tumor molecular profiling was analyzed using the FoundationOne CDx platform, 20 which analyzed base substitutions, indels, copy number alterations, and gene rearrangements across 324 cancer-related genes. Microsatellite instability and tumor mutational burden were also evaluated.
Results
Clinical Features
Patient 1: A woman in her early 50s presented with a 2 month history of gross hematuria, right-sided back pain radiating to her right knee, fatigue, and decreased appetite. An abdominal computed tomography (CT) scan revealed a solid 10.5 cm mass located in the lower pole of her right kidney, extending into the collecting system without evidence of vascular invasion or locoregional adenopathy. The patient underwent a laparoscopic right radical nephrectomy without complications and was discharged on the first postoperative day. She had no family history of tuberous sclerosis and was advised to undergo close follow-up.
Patient 2: A woman in her early 70s with a remote history of melanoma and chronic kidney disease (estimated glomerular filtration rate 44 mL/min/1.73 m2), presented with abdominal pain. She denied gross hematuria or flank pain. Magnetic resonance imaging revealed a 3.8 × 3.7 × 3.1 cm3 hyperenhancing exophytic mass with washout, arising from the anteromedial margin of the upper pole of the right kidney. The patient underwent a right radical nephrectomy without complications. She had a family history of multiple endocrine neoplasia type 1 (MEN-1), but genetic testing performed at ARUP Laboratories was negative. She also denied any family history of tuberous sclerosis.
Pathological Findings
Specimen 1: Gross examination revealed a well-circumscribed tumor measuring 10.2 × 9.0 × 8.0 cm3, occupying the entire lower pole of the kidney. The tumor was predominantly hemorrhagic and necrotic, with only small areas of viable yellow-tan tissue (Figure 1).
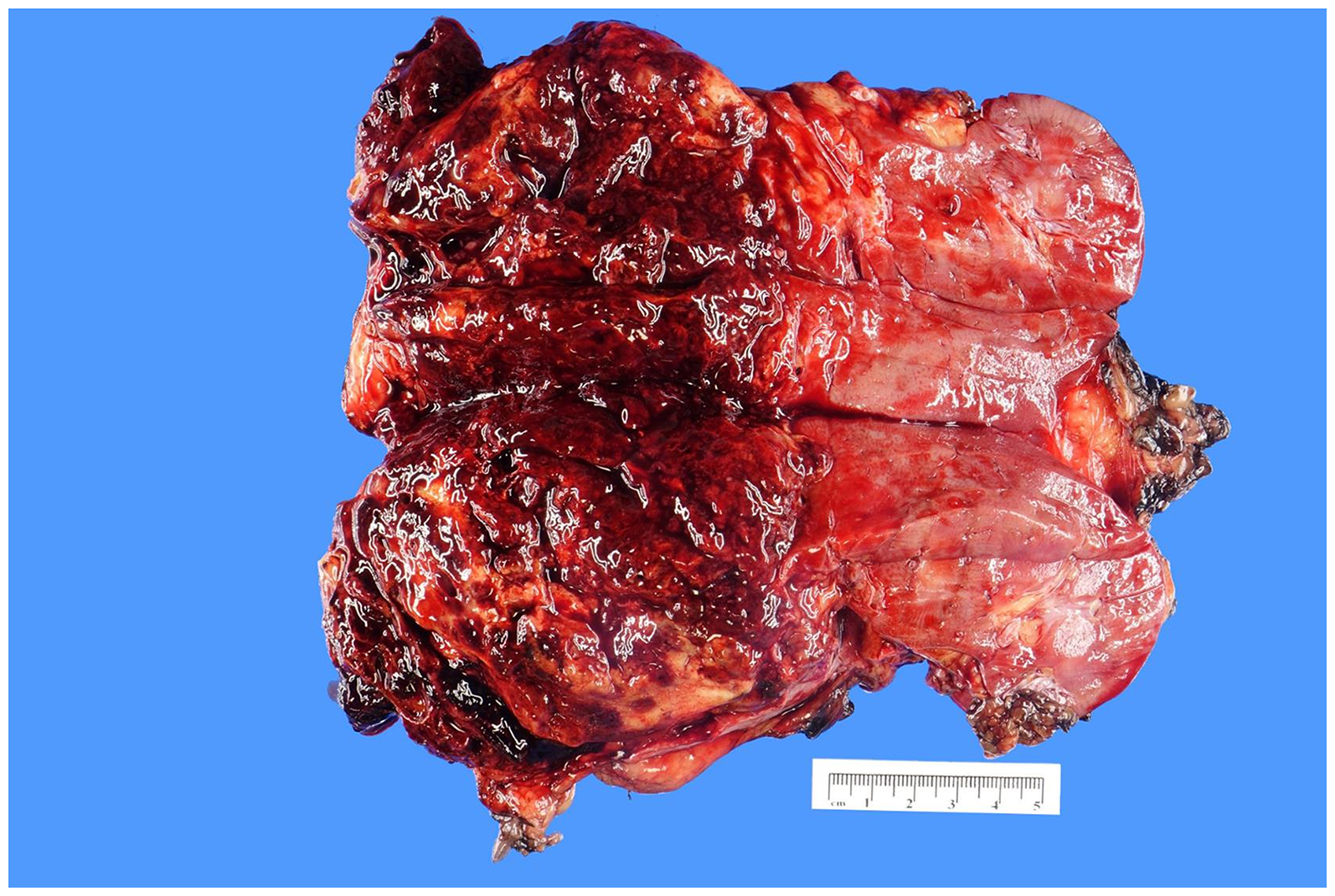
Gross photograph of a radical nephrectomy specimen showing a 10.2 × 9.0 × 8.0 cm3 tumor located in the lower pole of the kidney. The tumor was characterized by extensive hemorrhage and necrosis, with only minimal areas of viable tumor tissue.
Microscopically, the tumor consisted of nests of plump epithelioid cells and multinucleated giant cells with prominent nucleoli and abundant eosinophilic granular cytoplasm, atypical mitotic figures, and areas of tumor necrosis, all indicative of a predominantly epithelioid component (Figure 2). Notably, spindle cells and adipose tissue were absent. The tumor exhibited extensive hemorrhage with foci of hematopoiesis (erythroblasts) and golden-brown, coarsely granular, refractile hemosiderin pigments. Additionally, scattered black, finely granular pigments were identified and confirmed to be melanin.

Low-power H&E image showing smooth muscle cells radiating from thick-walled blood vessels. (B and C) High-power revealed plump epithelioid cells and multinucleated giant cells with prominent nucleoli and abundant eosinophilic granular cytoplasm, (D) atypical mitotic figures, and (E) areas of tumor necrosis. (F) Scattered foci of hematopoiesis (erythroblasts) were observed. (G and H) Golden-brown, coarsely granular, refractile hemosiderin pigments were visible. (I) Black, finely granular pigments.
Immunohistochemical staining demonstrated positivity for HMB45 and MART1, focal positivity for SMA, and rare positivity for S100. The tumor was negative for broad spectrum keratin AE1/3 and CAM5.2, PAX8, MiTF, and SF1. The presence of melanin was further confirmed by Fontana-Masson staining (Figure 3).

Immunohistochemical staining of the epithelioid angiomyolipoma demonstrated (A) positivity for MART1, (B) HMB45, and (C) focally positivity for SMA. The tumor cells were (D) negative for broad-spectrum keratin AE1/3 and CAM5.2 and (E) SF1. (F) The black, finely granular pigments were identified as melanin using the Fontana-Masson stain.
Specimen 2: Gross examination of the radical nephrectomy specimen revealed a 3.3 × 3.0 × 1.8 cm3 mass located at the hilum. The cut surface of the tumor was hemorrhagic and tan-yellow. The mass involved the soft tissue in the hilum and was located 0.2 cm from the renal vein margin. The adrenal gland was uninvolved.
Microscopically, the tumor consisted of epithelioid and plump spindle cells arranged in a diffuse growth pattern (Figure 4). Focal necrosis was present, surrounded by multinucleated giant cells and golden-yellow refractile hematoidin/hemosiderin pigments. The tumor cells exhibited prominent nucleoli and abundant eosinophilic granular cytoplasm, with scattered black, finely granular pigments. No atypical mitotic figures were identified.

(A) Low-power H&E image showing prominent, thick-walled blood vessels. (B) High-power revealed plump epithelioid cells. (C-E) Low- and high-power images showing golden-brown, coarsely granular, refractile hemosiderin/hematoidin pigments. (F) Black, finely granular pigments were identified as melanin. Immunohistochemical staining showed that the epithelioid angiomyolipoma was (G) positive for MART1 and (H) focally positive for HMB45. Abbreviation: H&E, hematoxylin and eosin.
Immunohistochemical staining showed that the tumor cells were diffusely positive for MART1 and focally positive for HMB45. The tumor was negative for CAM5.2 and CA9. It should be noted that S100 and SOX10 staining was not performed because the patient's melanoma was resected from her forehead 26 years prior to her current specimen. There was no clinical concern for recurrence and thus it was highly improbable to be metastatic melanoma. Additionally, an incidental cystic clear cell papillary renal cell tumor, measuring 3.0 cm, was identified in the background. The surgical margins were uninvolved.
Molecular Findings
The molecular analysis of specimen 1 revealed the following details: The tumor's mutational burden was 0 mutations per megabase, and its microsatellite status was stable. Notable molecular alterations included mutations in TSC2 (K500fs*20 and H1746_R1751del) and TP53 (R273C).
Follow-Up
Patient 1: There was no evidence of residual disease upon abdominal CT scan 4 months postradical nephrectomy.
Patient 2: The patient had stable renal function and no evidence of recurrence 10 years postsurgery.
Discussion
As rare mesenchymal tumors, PEComas are thought to originate from perivascular epithelioid cells. These tumors are characterized by fascicles of spindle cells or sheets of epithelioid cells expressing both smooth muscle and melanocytic markers. Epithelioid AML, a well-recognized subtype, is defined in the 2016 WHO Classification as a rare variant of AML comprising at least 80% epithelioid cells. 21 Morphologically, epithelioid AMLs may present in 1 of 2 patterns: a “carcinoma-like” appearance, demonstrated by specimen 1 in our study, or a pattern featuring epithelioid and plump spindle cells in diffuse growth, such as specimen 2. 22
The potential aggressiveness of epithelioid AMLs is well-established, though the clinicopathological features predicting clinical behavior remain debated.23,24 In a 2018 literature review, Zhan et al identified 8 malignant features in epithelioid AML. 25 They proposed that tumors meeting at least 5 of the following criteria may be considered malignant: size ≥5 cm, metastasis, infiltration, necrosis, ≥50% atypical epithelioid cells, cytologic atypia, atypical mitotic figures, and vessel invasion. Specimen 1 in our study met 5 of these criteria: large size, infiltration, necrosis, cytologic atypia, and atypical mitotic figures, while specimen 2 only met criteria for cytologic atypia and necrosis. Specimen 2's low potential for malignancy was reflected by no signs of recurrence 10 years after surgery. Although specimen 1 can be considered malignant per Zhan et al's criteria, it was diagnosed more recently and thus does not have follow-up information further than 4 months postop, at which point there was no radiologic evidence of recurrence.
The true prevalence of pigmented renal epithelioid AMLs is likely obscured by the challenge in distinguishing it from melanoma, pigmented paraganglioma, and pigmented renal cell carcinoma. Melanomas exhibit a spectrum of morphologic patterns and test positive for melanocytic markers, similar to PEComas. Melanomas can be identified by positive staining for S100 and negative staining for smooth muscle markers, which is uncommon in PEComas. Conversely, pigmented paragangliomas are positive for neuroendocrine markers (chromogranin A and synaptophysin) and S100 but negative for melanocytic markers.26,27
Pigmented renal cell carcinomas are positive for keratins and negative for melanocytic markers. However, melanocytic markers may be present in MiT family translocation renal cell carcinomas, which involve TFE3 and TFEB rearrangements. These can be differentiated from epithelioid AML through the use of PAX8 and CD68 (PGM1) markers, which are negative and positive in epithelioid AML, respectively, and show opposite staining patterns in MiT family translocation renal cell carcinomas.28,29
Similar to these molecularly defined renal cell carcinomas, Xp11 translocations, which can facilitate rearrangements of the TFE3 gene, have also been identified in malignant epithelioid AMLs. 30 This has led to variations in terminology such as “Xp11 PEComa,” and “melanotic Xp11 renal neoplasm. 22 ” Melanotic Xp11 translocation renal cancers are now classified as “pigmented TFE3-rearranged PEComas” when they are negative for renal tubular markers such as PAX8 and diffusely positive for melanocytic markers and cathepsin K. 31
While classic AMLs are strongly associated with tuberous sclerosis, 32 only 2 of the 16 pigmented epithelioid AML tumors reported occurred in patients with a history of tuberous sclerosis (Table 1). One of our pigmented epithelioid AML tumors revealed notable molecular alterations, including mutations in TSC2 (K500fs*20 and H1746_R1751del). TSC2 mutations have been identified in approximately 34% of patients with renal AML. 33 Furthermore, TSC2 mutations have been detected from epithelioid AML specimens in patients with no history or clinical signs of tuberous sclerosis in studies from as far back as 1998. 34 Disruption of the TSC1/2 complex in PEComas is associated with increased activation of the mammalian target of rapamycin (mTOR) signaling pathway, thereby promoting cell growth. 35 Consequently, malignant PEComa patients harboring a TSC2 mutation tend to respond more favorably to mTOR inhibitors than those without the mutation. 36 Although promising, these studies are limited in sample size and the prognostic significance of TSC2 alterations in PEComas warrants further study.
The other molecular alteration found in specimen 1 was TP53 (R273C), which is a missense mutation that has been shown to enhance proliferation and invasion of in vitro cancer cell lines through loss of the tumor suppression function of p53 and subsequent decrease in apoptosis. 37 TP53-mutated renal and hepatic AMLs have been associated with aggressive epithelioid features.38–44 For example, 1 study identified TP53 mutations in 3 out of 8 epithelioid AML tumors, whereas none were found in 8 classic AML tumors. 45 However, the prognostic significance of TP53 alterations in PEComas remains unclear and warrants further investigation.
Diagnosing pigmented epithelioid AMLs remains challenging, as an accurate diagnosis continues to rely on a meticulous search for melanin pigment and comprehensive immunohistochemical analysis. The clinical implications of pigmentation in epithelioid AML are still uncertain due to the scarcity of reported tumors. Our findings highlight the critical role of these diagnostic approaches in addressing this exceptionally rare condition. To enhance characterization and understanding of epithelioid AMLs, future multi-institutional studies with larger patient cohorts and prospective tissue collection are required.
Footnotes
Ethical Approval and Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data used in this case report is available per the manuscript.
Trial Registration
Not applicable, because this article does not contain any clinical trials.