
Abstract
Prediction of the biological potential of a tumor is the fundamental goal of the histomorphological, immunohistochemical, and molecular genetic assessment in any sample of human neoplasia. For some tumors, the risk of clinically aggressive behavior is implicit in the classification itself; for other tumors, there are specific attributes, histological or otherwise, that portend a better or worse prognosis. These outcome-associated characteristics often drive key treatment decision making, particularly in an era of therapy that is increasingly accessible, frequently targeted, and generally more efficacious. In the case of dermatofibrosarcoma protuberans (DFSP), potential for aggressive tumor behavior is heralded by what was first observed as a morphological transformation manifesting as a “fibrosarcomatous” appearance: a cellular tumor component arranged in distinctive fascicles exhibiting more conspicuous mitotic activity. In this article, we review the criteria for and clinical implications of diagnosing fibrosarcomatous DFSP. We highlight the potential for histological variation and heterogeneous outcomes, suggesting that fibrosarcomatous DFSP is not a monolithic entity. Finally, we summarize recent advances in our understanding of the molecular correlates of fibrosarcomatous transformation, discuss the diagnostic and treatment-related implications of these advances, and propose future directions for gaining further insight into fibrosarcomatous DFSP, its defining features, and therapeutic vulnerabilities.
Keywords
Introduction
The primary aim of the histopathological assessment of soft tissue tumors is to evaluate the biological potential of the tumor in an effort to accurately guide treatment—optimizing therapeutic decision making with curative intent, while minimizing treatment-related morbidity. For some soft tissue neoplasms, the tumor classification and stage together represent adequate information to guide management; in these tumors, the biological potential is implied by the classification. For another subset of soft tissue tumors, there is a spectrum of clinical behavior that correlates to some degree with a morphological continuum, leading to the development of grading systems that can be used to estimate malignant potential. These histological grading schemes, such as the Fédération Nationale des Centres de Lutte Contre Le Cancer (FNCLCC) system, tabulate histological features associated with risk of clinically malignant behavior.
For a smaller subset of soft tissue neoplasms, there are distinctive histopathological transitions that occur as tumors assume a more aggressive clinical phenotype, warranting a tumor-specific diagnostic approach to the identification of this apparent form of oncogenic progression. In myxoid liposarcoma, for example, the tumor transitions, either gradually or abruptly, to a high-grade “round cell” phenotype coinciding with greater potential for metastatic spread and worse overall survival. An analogous transition occurs in a subset of dermatofibrosarcoma protuberans (DFSP) wherein the neoplasm adopts a constellation of histological features historically perceived as being indicative of a “fibrosarcoma”—namely, a more densely cellular, fascicular appearance. While it is clear that fibrosarcomatous (FS) transformation does occur in a small fraction of DFSP, and that this histopathological evolution correlates to some degree with clinically aggressive tumor behavior, FS-DFSP remains a practical challenge because of its rarity, rather subjective distinguishing diagnostic features, uncertain molecular underpinnings, and potentially significant implications for prognostication and treatment. Here, we aim to provide practical guidance for current diagnostic approaches to FS-DFSP, and to build a foundation for further investigation of this enigmatic phenomenon.
Epidemiology
FS transformation occurs in 3% to 21% of DFSPs, depending on the series.1–12 The median age at diagnosis for FS-DFSP is typically cited as being higher than that for conventional DFSP, although the reported ranges of median age at presentation varies considerably (37-45 years for DFSP and 43-56 years for FS-DFSP).8,9,13–17 However, some studies have not demonstrated significant differences in age between these groups.8,10,11 While some studies have pointed to an increased risk of FS transformation in women, a large series of DFSPs showed no evidence for a difference in sex of patients with FS-DFSP.5,8,11,13,14 Although the incidence of DFSP in black patients is almost double that among white patients, there is no evidence that tumors in black patients undergo FS transformation at a different rate.2,10,15–19
Clinical Presentation and Predisposing Factors
In comparison with conventional DFSP, FS-DFSP presents with pain, rapid enlargement, and larger tumor size.8,13,20–24 Evidence on whether FS transformation is more common in recurrent versus primary tumors is mixed.8,25–27 DFSPs at all anatomic sites appear equally likely to undergo FS transformation.10,13,21,25,28,29 However, head and neck and acral sites have higher rates of recurrence, possibly due to the difficulty in achieving margin-negative (R0) resections.
Trauma is a hypothesized predisposing or inciting factor for DFSP formation itself.28,30–39 Whether trauma predisposes to FS transformation is much less well documented. One study found a history of trauma significantly more often in FS-DFSP than DFSP. However, another study that examined the question did not reproduce this finding.10,13 A third large cohort study that included trauma history found no relationship with FS transformation. 30 Similarly, while radiation has been reported as an antecedent history in DFSP, there is no evidence that it is associated with increased risk of FS transformation.40,41
Special Populations
Although rapid growth of many tumors, including DFSP, has been observed during pregnancy, the percentage of tumors undergoing FS transformation during pregnancy does not appear to be increased.42–44
FS transformation is exceptionally uncommon in children.45–48 Transformed tumors in children behave in the same aggressive fashion as in adults and require aggressive treatment.45,46 In children, certain forms of congenital immunodeficiency, especially adenosine deaminase-deficient severe combined immunodeficiency (ADA-SCID), predispose to multiple DFSPs, presumably through DNA breaks. While this mechanism is postulated to lead to additional DNA damage and increased risk of FS transformation, studies have not confirmed this hypothesis, although follow-up notably has been limited.49,50 It is also somewhat unclear if this association with DFSP is related to the immunodeficiency in these patients, especially in light of limited evidence that DFSP has any association with other immunocompromised states. Interestingly, the typical supernumerary ring chromosomes found in most adult DFSP are less common in children. Pediatric DFSPs more often show balanced or unbalanced translocations and may be a somewhat different disease.48,51–53
The evidence that acquired immunodeficiency predisposes to DFSP is limited, and the effect on the risk of FS transformation is unclear.54–59 Anecdotally, 2 patients taking the TNF-alpha blocker adalimumab were reported to develop FS-DFSP.55,60
Histopathology, Diagnostic Criteria, and Variant Morphologies
The diagnosis of FS-DFSP is made when the typical storiform pattern of DFSP is replaced by fascicular, often herringbone, architecture (Figure 1). The fascicles are densely cellular, and the neoplastic cells typically show mildly increased atypia and markedly increased mitotic index (Figure 2).25,51,52,61 A cutoff for what constitutes enough mitotic activity for a diagnosis of FS-DFSP has not been established and reported mitotic indices vary widely in the literature. However, in general, most conventional DFSP show 0-5 mitotic figures per 10 high-power microscopic fields, while most FS-DFSP show >5 mitoses per 10 high-power microscopic fields, although cutoffs of 10 mitotic figures per 10 high-power fields have been used in some studies.10,12,27,62,63
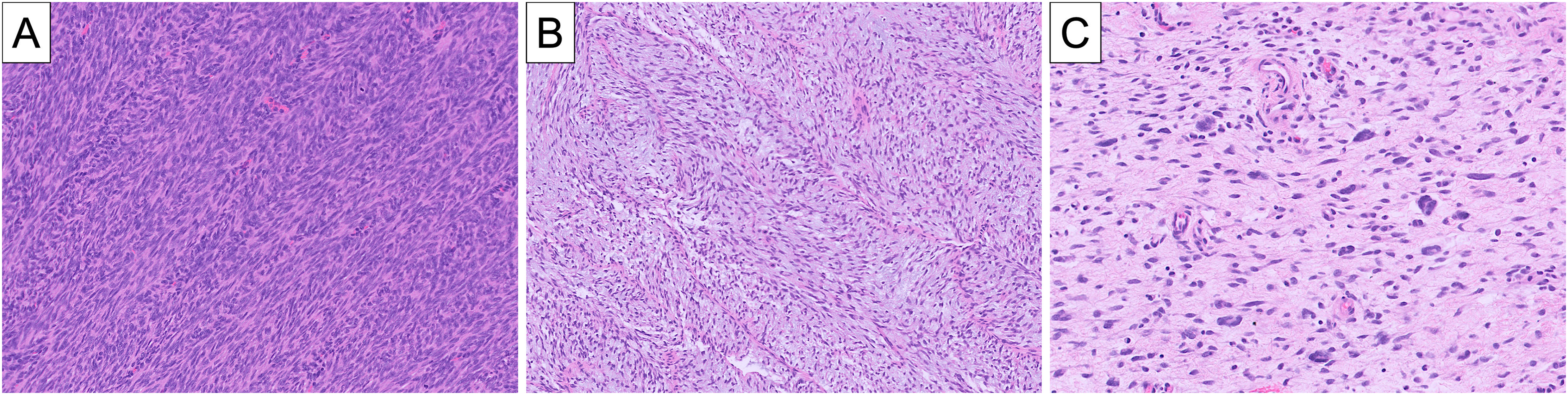
Fibrosarcomatous dermatofibrosarcoma protuberans (DFSP) can exhibit a wide range of morphology, even within a single tumor: the typical hypercellular appearance with herringbone conformation (A), a looser and less cellular arrangement of fascicles with myxoid stroma (B), and a distinctively disorganized distribution of spindle cells with unusually misshapen and pleomorphic nuclei (C).

Comparison of conventional (A) and fibrosarcomatous (B) areas of a dermatofibrosarcoma protuberans (DFSP). While the 2 components demonstrate similar cellularity, the conventional component has storiform architecture and no significant mitotic activity, whereas the fibrosarcomatous component has fascicular architecture and conspicuous mitotic figures.
Areas of FS morphology are more often seen in the deep portions of the tumor25,27,33,64 and may show either abrupt or gradual transition from areas of conventional DFSP.51,52,65,66 Generally, a cutoff of 5% of the tumor comprising fascicular growth has been regarded as sufficient for the diagnosis of FS-DFSP,20,67 although any amount of fascicular growth should be noted and may be an indication for additional clinical intervention and possibly further tissue sampling if the tumor is not entirely removed or submitted for histological evaluation. This largely historical cutoff has been validated by the reported occurrence of metastasis of a DFSP with only 5% FS component. 63
In evaluating variant DFSPs lacking extensive storiform growth (myxoid, atrophic, and giant cell fibroblastoma), the diagnosis of FS transformation rests on finding fascicular growth with increased cellularity (Figure 3). While mitotic activity is generally increased, mitotic activity alone is not sufficient for a diagnosis of FS-DFSP. However, increased mitotic index without FS transformation has been independently associated with poor outcomes.6,25 For the converse situation, fascicular growth without increased mitotic activity, there is little evidence or consensus on whether this is sufficient for the diagnosis of FS-DFSP.68,69

A subset of conventional and fibrosarcomatous dermatofibrosarcoma protuberans (DFSPs) will exhibit prominent myxoid stroma. Fibrosarcomatous DFSPs with myxoid stroma will retain their characteristic fascicular or herringbone architecture (A), as highlighted by CD34 immunostaining in this tumor (B). Myxoid stroma in the setting of a rich tumor blood supply composed of narrow-caliber, curvilinear vessels has the potential to mimic myxofibrosarcoma (C).
While certain molecular subtypes are tied to altered risk of FS transformation and aggressive behavior (see Molecular pathology below), myxoid and pigmented (Bednar) histological variants have similar clinical outcomes.25,70–72 Giant cell fibroblastoma and atrophic DFSP appear to have lower rates of FS transformation, although whether that is due to the histological variant or the fact that they are more common in children is unclear.20,31,73–75 Myoid differentiation or myoid nodules are observed preferentially, but not exclusively, in FS-DFSP. Myoid nodules are not thought to be independent risk factors outside of FS transformation.76–80
Despite the fact that the vast majority of high-grade sarcomas arising within DFSP show typical, monomorphic fibrosarcoma morphology, rare examples of pleomorphic sarcoma have been reported.81–83
Immunohistochemistry
Several immunohistochemical markers are preferentially expressed or lost in FS-DFSP compared to conventional DFSP. While these can add some ancillary support for the diagnosis, the diagnosis ultimately rests on the classic morphological findings of fascicular growth, increased cellularity, and increased mitotic activity.
Absent or decreased reactivity for CD34 is seen in approximately half of FS-DFSP (Figure 4), although this estimate ranges wildly in the literature from 11% to 100%.4,8,9,14,25,63,66,84,85 A quantitative study using computerized image analysis established 76% sensitivity and 95% specificity for FS transformation based on reduced CD34 staining. 86 PReferentially expressed Antigen in MElanoma (PRAME) has also emerged as a potential marker of FS transformation; digital spatial profiling revealed significant upregulation of PRAME RNA expression in FS-DFSP. Subsequent IHC (immunohistochemistry) studies showed moderate to strong PRAME reactivity in >50% of nuclei in 70% of FS-DFSP compared to only 4% of conventional DFSP. 87 S100 protein, while almost always negative in DFSP, has been reported to be positive in rare molecularly confirmed examples of DFSP with a propensity for FS transformation. 88

Fibrosarcomatous dermatofibrosarcoma protuberans (DFSP) exhibiting characteristic fascicular architecture and hypercellularity (A), as well as diffuse loss of CD34 expression by immunohistochemistry (B). Positive CD34 immunostaining in vascular endothelium serves as an internal positive control.
Not surprisingly, given the increased mitotic activity associated with the diagnosis, Ki-67 is consistently elevated in FS-DFSP (18%-42%) compared to DFSP (8%-10%).8,13,23,89 p53 labeling has also been noted to be higher in FS-DFSP than in DFSP, likely correlating with additional mutations within the p53 pathway.13,20,85,90–92 Loss of p16 or RB1 has been proposed as a potential marker of FS-DFSP (Figure 5), but loss has also been seen in conventional DFSP, limiting its utility (see Molecular pathology below).93,94 Despite the associations that FS-DFSP has with these IHC features, we do not routinely rely on IHC for the diagnosis of FS transformation, anchoring the diagnosis instead on the defining morphological characteristics.
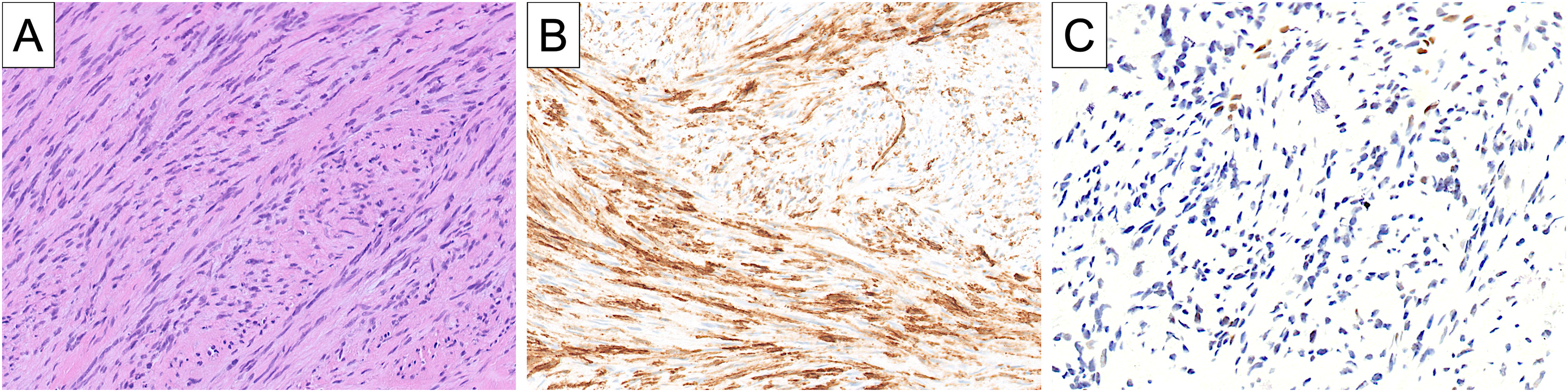
Representative example of fibrosarcomatous dermatofibrosarcoma protuberans (DFSP) (A) exhibiting partially diminished CD34 expression (B) and loss of RB1 expression (C). A pathogenic RB1 alteration was confirmed by molecular genetic testing in this tumor.
Molecular Pathology
Conventional DFSP is driven by a recurrent reciprocal translocation, t(17;22)(q22;q13), and a resulting supernumerary ring chromosome. This translocation links the COL1A1 gene on chromosome 17q21–22 with the PDGFB gene on chromosome 22q13, resulting in the juxtaposition of PDGFB with the constitutively active promoter of COL1A1. Overexpression of PDGFB at the mRNA level leads to increased production of the PDGFB growth factor, which is transmitted in an autocrine or paracrine manner to stimulate oncogenic signaling pathways, such as RAS/MAPK, PI3 K/Akt, and JAK/STAT.
In FS-DFSP, the COL1A1::PDGFB fusion is present in both the conventional and FS components, confirming their clonal genetic relationship and shared cellular origin. 95 Employing paired analyses of conventional and FS areas of DFSPs, it has been observed that the FS component exhibits low-level copy number gains of the pathogenic PDGFB fusion (median COL1A1::PDGFB copy gain 2.8, range 0-5.9) in the majority of tumors, although this copy number gain does not appear to be necessary for FS transformation insofar as it is not seen in every transformed tumor. 96 While the level of copy number gain is low in comparison to highly amplified genes such as MDM2 in well-differentiated/dedifferentiated liposarcoma and MYCN in neuroblastoma, it has been suggested that the oncogenic stimulus generated by the COL1A1::PDGFB fusion is sufficiently strong that even relatively modest gains can yield strong growth-promoting effects. Indeed, analysis of PDGFB expression in DFSP by RNA in situ hybridization has demonstrated stronger expression in FS-DFSP relative to conventional DFSP (Figure 6). 97

In the large majority of dermatofibrosarcoma protuberans (DFSPs), the COL1A1::PDGFB fusion drives overexpression of PDGFB, which can be visualized by RNA in situ hybridization. Although PDGFB overexpression is evident in both conventional (A, B) and fibrosarcomatous (C, D) tumor components, fibrosarcomatous transformation often coincides with increased PDGFB expression relative to the conventional component.
FS transformation often coincides with genomic alterations beyond the COL1A1::PDGFB fusion itself: approximately 75% of FS-DFSPs harbor at least one non-PDGFB/PDGFD-related genetic alteration, compared to about 24% of conventional tumors. 98 In particular, FS variants are more than 4-fold as likely as conventional DFSP (45% vs 9.5%) to demonstrate an SNV (single nucleotide variant) or indel. Along these lines, it has been shown that FS-DFSP is characterized by increased genomic amplifications or deletions (mean: 27.2 regions per sample) relative to conventional DFSP (mean: 7.7 regions per sample). 11 Correlating with these broad molecular hallmarks of transformation, conventional DFSP generally is characterized by a lower tumor mutational burden (TMB) relative to transformed tumors (median 1 mut/MB for conventional vs 2 mut/MB for FS). 98 Taken together, these genetic features imply that the morphological FS transformation of DFSP is strongly associated with a corresponding genomic transformation.
As a key tumor suppressor, p53 has been implicated in the pathogenesis of FS transformation by several studies. In their analysis of more than 40 DFSPs with FS areas, Abbott et al 27 found that p53 was expressed by immunohistochemistry in 92% of FS components and only 3% of conventional components. Other authors also have observed p53 expression in FS-DFSP relative to conventional tumors, 13 but in a much smaller proportion of tumors. This immunopositivity for p53 in FS-DFSP correlates with an increased incidence of TP53 pathogenic alterations, or mutations affecting other components of the p53 pathway, relative to conventional DFSP. MDM2, which acts in part via suppression of p53, also has been shown to be overexpressed in a FS-DFSP with chromosome 12q amplification as well as CDKN2A/B deletion limited to the FS component. 99 In another series of FS-DFSP, TP53 alterations were relatively infrequent, limited to one FS-DFSP with distinctive pleomorphic cytomorphology and extensive loss-of-heterozygosity. 98 Indeed, in contrast to aforementioned studies, Li et al found “negative” (wild-type) p53 staining in all 24 FS-DFSPs and 195 conventional DFSPs. 8 Thus, while it appears that TP53 alterations may be associated with FS transformation, they likely account for only a subset of FS-DFSP.
Loss of tumor suppressor function with FS transformation in DFSP is not limited to p53. More frequently, broad genomic profiling reveals alterations in the NF1 gene encoding neurofibromin, a tumor suppressor implicated in the pathogenesis of nerve sheath tumors. In one study of FS-DFSP, 3 of 31 tumors harbored a pathogenic NF1 alteration. 98 DFSP has been reported once in a patient with neurofibromatosis, however this tumor was not observed to exhibit FS histology; the tumor was treated with wide local excision and did not recur. 100 Both PDGFB/PDGFRB and NF1 act, at least in part, via RAS/MAPK and PI3/Akt signaling pathways, suggesting that co-alteration of NF1 in DFSP may serve to potentiate the oncogenic effects of PDGFB. NF1 also impacts signaling of other receptor tyrosine kinases, raising the possibility that treatment with somewhat broadly targeting tyrosine kinase inhibitors, such as imatinib mesylate, may be a useful strategy in the event of combined NF1 alteration and COL1A1::PDGFB fusion. 100
Other tumor suppressors exhibiting loss of expression or function in FS-DFSP include CDKN2A/B which encodes both p16INK4a (p16) and p14ARF. CDKN2A/B loss is a recurrent phenomenon across human malignancies, and represents an indicator of poor prognosis among soft tissue sarcomas in particular. 101 In select examples of FS-DFSP, CDKN2A/B loss has been confirmed by FISH (fluorescence in situ hybridization), along with further confirmation of loss of p16 expression by immunohistochemistry.11,94,98,99,102 Even more notably, deletions of CDKN2A/B may correlate with metastatic behavior 5 and imatinib resistance, 94 although CDNK2A/B loss itself may not be entirely specific for FS transformation among DFSPs. Indeed, DFSP has been reported, in conjunction with multiple melanomas, in a patient with germline CDKN2A/B mutation, although this DFSP was not described as having features of FS transformation. 103 Recurrent oncogenic alterations associated with FS transformation in DFSP are not limited to loss-of-function of tumor suppressors. For instance, noncoding mutations affecting the TERT promoter, seen in a diverse array of human tumors, are also observed in a subset of FS-DFSP, resulting in increased telomerase expression, which promotes cell proliferation and survival. 98
While the vast majority of DFSPs, including those with FS transformation, are primarily driven by COL1A1::PDGFB, tumors are rarely associated with an alternative fusion, most notably involving the platelet-derived growth factor D chain (PDGFD). These PDGFD-rearranged DFSPs more often manifest as subcutaneous masses with relatively circumscribed borders in comparison to their more superficial and more infiltrative PDGFB-related counterparts.104,105 Further contrasting with PDGFB-rearranged DFSPs, the PDGFD translocation involves distinctive partner genes, namely COL6A3, EMILIN1, EMILIN2, or TNC.105,106 PDGFD-related DFSPs were initially observed to harbor the most common COL6A3 gene partner, exhibiting a distinctive predilection for the chest wall or breast of female patients. 107 These DFSPs with COL6A3::PDGFD fusions generally exhibit conventional histology with rare reported FS-DFSPs among this subtype; however, FS features appear to be much more commonly encountered in EMILIN2::PDGFD-rearranged DFSPs, the second most common genetic subtype of PDGFD-associated DFSP. FS features have been reported in up to as many as 75% of these EMILIN2::PDGFD DFSPs, which also tend to occur more commonly in males.102,106 FS transformation has also been observed in the even rarer EMILIN1- and TNC-related PDGFD-rearranged DFSPs,106,108 although very few tumors with these molecular characteristics have been reported to date. Collectively, these observations indicate that a subset of DFSPs with FS transformation is associated with uncommon variant oncogenic fusions, especially EMILIN2::PDGFD.
Molecular changes coinciding with FS transformation are not limited to genetic alterations but instead may reflect epigenetic changes that promote cell proliferation, survival, and metastatic spread. For instance, altered signaling via the Akt-mTOR pathway has been demonstrated in FS-DFSP relative to conventional DFSP, and may represent a targetable susceptibility in imatinib-resistant tumors.89,109 Broader analyses of molecular pathways activated during the course of FS transformation implicate interactions with the extracellular matrix, as well as molecular signaling features related to cell adhesion/migration and “epithelial-mesenchymal transition,” but notably no evidence of changes in the adaptive immune response.11,87 It is tempting to consider that the evident changes in these pathways account, at least in part, for the distinctive architecture and potential for metastatic spread that characterizes FS-DFSP. Intriguingly, as noted above, the most highly upregulated gene in FS-DFSP relative to conventional DFSP in one broad transcriptomic profiling study was PRAME. 87 Validation of this finding by immunohistochemistry showed that PRAME was positive for expression in 7 of 10 FS-DFSP (70%) and only 1 of 23 conventional DFSP (4%), confirming an earlier study that showed PRAME positivity by immunohistochemistry is 86% sensitive and 90% specific for the diagnosis of FS-DFSP relative to conventional DFSP. 86 Thus, epigenetic features that distinguish FS-DFSP from conventional DFSP include gene expression changes that may be diagnostically or therapeutically relevant.
Prognostic Significance, Grading, and Staging
Across studies, FS-DFSP shows markedly more aggressive behavior than conventional DFSP. Local recurrence and metastatic rates are consistently higher for FS-DFSP (see Table 1). However, regarding published studies of outcomes for FS-DFSP, there are notable exceptions. One series of 18 FS-DFSPs treated with wide local excision reported by Goldblum et al 66 did not have any metastases with 81.5 months of follow up. The Castle et al’s 19 2013 study of patients treated with conservative surgery and radiation had excellent outcomes with disease-free survivals of 98% and 93% at 5 and 10 years without any prognostic difference between DFSP and FS-DFSP.
Primary Series Comparing Outcomes of DFSP and FS-DFSP.
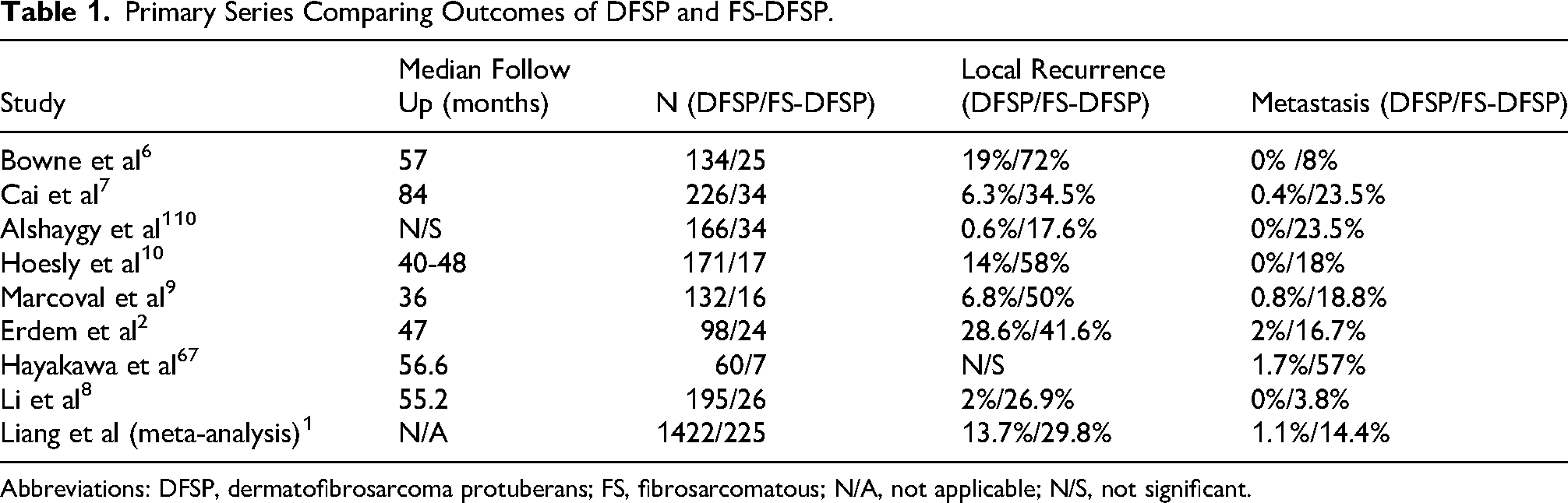
Abbreviations: DFSP, dermatofibrosarcoma protuberans; FS, fibrosarcomatous; N/A, not applicable; N/S, not significant.
Grading of FS-DFSP is controversial, but is advisable given the observation that mitotic index, and possibly necrosis, are independently correlated with outcome. Within the FNCLCC system, authors have assigned differentiation scores of both 2 and 3.12,110 Necrosis in FS-DFSP is rare (Figure 7), which limits the ability to establish its clinical significance. 6 Several studies have shown no clinical significance.25,27 Another analysis showed that necrosis in FS-DFSP was linked to significantly decreased overall survival, but was not an independent risk factor for poor survival upon multivariate analysis. 2 Given the rarity of pleomorphic sarcoma arising in DFSP, reliable data on the significance of cytological pleomorphism/atypia is not available, but highly aggressive clinical behavior has been reported. 25 One study found that the extent of FS transformation within a tumor conferred higher risk of recurrence or metastasis. 2 Other studies, however, have not found an association between the extent of FS morphology and clinical behavior.11,22,25,27

Unlike conventional dermatofibrosarcoma protuberans (DFSP) (A), fibrosarcomatous transformation (B) often imparts a hypercellular appearance with somewhat larger, subtly more pleomorphic nuclei with coarser chromatin. Mitotic activity is almost always increased in the fibrosarcomatous component. More uncommonly, there is geographic tumor necrosis in fibrosarcomatous DFSP (C), here imparting a peritheliomatous pattern to the residual viable tumor.
The College of American Pathologists (CAP) considers FS-DFSP appropriate for American Joint Committee on Cancer (AJCC) staging. Musculoskeletal Tumor Society (MSTS) staging systems have also been applied to DFSP and FS-DFSP.111,112 Additional specific staging systems for FS-DFSP have been proposed as well. 112
Differential Diagnosis
In most instances, the diagnostic challenge with respect to FS-DFSP is to reliably identify it in a background of known DFSP. Examples of pure FS-DFSP are exceedingly rare, but may raise a differential diagnosis of other high-grade monomorphic spindle cell sarcomas such as synovial sarcoma, malignant peripheral nerve sheath tumor, and other tyrosine kinase receptor-associated sarcomas, particularly in the metastatic setting (Figure 8). Conventional immunohistochemistry is largely unhelpful in this differential diagnosis as TLE1, pan-Trk, and trimethylated histone H3K27 all lack specificity to some degree.113–115 In particular, pan-Trk can be expressed in DFSP, leading to potential misdiagnosis as an NTRK-rearranged neoplasm (Figure 9). However, emerging “next generation” immunohistochemical and RNA ISH markers for SS18::SSX fusion, SSX C-terminus, and PDGFB RNA ISH (in situ hybridization) can be helpful.97,116,117 STAT6 immunohistochemistry is also useful in distinguishing FS-DFSP from solitary fibrous tumor, insofar as FS-DFSP can show diffuse and strong CD34 expression and develop “hemangiopericytoma-type” thin-walled, branching blood vessels (Figure 10). Clinical context remains key to accurately assessing a differential diagnosis that encompasses FS-DFSP and histologically related entities—in most metastatic FS-DFSPs, patients either will present with a large dermal or subcutaneous primary mass, or will present with recurrent or neglected disease. Lastly, targeted molecular genetic testing represents a critical tool in deciphering this differential diagnosis as the finding of a characteristic DFSP-associated fusion can provide strong support for the diagnosis of DFSP, along with a justification for targeted therapy.

Because of their dense cellularity, spindled cytomorphology, plump nuclei, and fascicular architecture, fibrosarcomatous dermatofibrosarcoma protuberans (DFSPs) (A and B), have the potential to mimic malignant peripheral nerve sheath tumor or synovial sarcoma, both of which seldom present as superficial masses in the way that DFSP ordinarily manifests. On the other hand, fibrosarcomatous DFSP metastasizing to visceral sites usually will be accompanied by a history of previously treated or neglected tumor at the primary site.

This fibrosarcomatous dermatofibrosarcoma protuberans (DFSP) (A) was mistaken for an NTRK-rearranged spindle cell neoplasm owing to patchy expression of S100 protein (B) and weak-moderate expression of pan-Trk (C). Evidence of diffuse PDGFB expression by RNA in situ hybridization (D) helped to confirm the correct diagnosis.

Fibrosarcomatous dermatofibrosarcoma protuberans (DFSP) can develop “hemangiopericytoma”-type vasculature (A). This feature, in combination with persistent strong CD34 expression in a subset of fibrosarcomatous DFSPs, can lead to histological mimicry of solitary fibrous tumor.
Management and Therapeutic Considerations
Surgery with widely negative margins is the mainstay of therapy for DFSP and especially for FS-DFSP. Recurrence rates for closely excised tumors skyrocket to 100% in some series of FS-DFSP compared to 28% for DFSP. 6 Furthermore, one study found that successful margin-negative (R0) resection reduced the risk of metastasis to 0% over a 5 to 17 year follow-up interval. 66 The growing trend of performing Mohs surgery for DFSP in order to guarantee a negative margin is controversial in FS-DFSP due to the many Mohs layers typically needed and the size of the defect created.20,21
Imatinib is a mainstay of therapy for unresectable DFSP. Initial therapy of FS-DFSP with imatinib is generally successful with partial responses of 50% to 80%.1,20,118–122 However, the response is less durable in FS-DFSP than in DFSP, likely due to the emergence of resistance from increased genomic instability.14,51,89,94,118,120,121,123,124 Imatinib-resistant FS-DFSP may respond to other tyrosine kinase inhibitors such as sunitinib, pazopanib, sorafenib, and apatinib, indicating potentially promising new strategies for targeted therapy in FS-DFSP, likely reflecting distinctive suscpetibilities.5,31,74,94,119,121,123,125–129
Radiation therapy is employed primarily in the adjuvant setting, but can also be used as primary treatment and in palliative settings. DFSP and FS-DFSP are considered radiosensitive tumors.20,54,63,111 However, it is unclear if FS-DFSP is more radiosensitive. 3 High local control rates of pooled cohorts of DFSP and FS-DFSP patients treated with wide local excision and radiation therapy suggest, however, a possible benefit. 19
Conclusions and Future Directions
The accurate diagnostic classification of FS-DFSP is key to appropriately risk-stratifying and adequately treating these rare malignancies. On the one hand, FS-DFSP must be distinguished from conventional DFSP, which harbors significantly less potential for metastatic spread, while on the other hand, FS-DFSP should not be mistaken for other relatively monotonous spindle cell sarcomas, such as synovial sarcoma or MPNST. Morphology remains the cornerstone of diagnosis insofar as FS transformation is largely defined by its fascicular architecture, typically increased mitotic activity, and variably pronounced cytological atypia. The rarity of FS-DFSP, qualitative nature of the diagnostic criteria, variability of the histomorphological features, and significant potential impact of the diagnosis together can make the diagnosis of FS-DFSP a challenge and warrant expert consultation for at least a subset of such tumors.
Support for the diagnosis can come in the form of ancillary immunohistochemical testing for CD34, which tends to be lost in a subset of FS-DFSP, although CD34 loss is neither entirely sensitive, nor completely specific, for the distinction of FS-DFSP from conventional DFSP. Conversely, in the context of a monomorphic spindle cell sarcoma, significant CD34 expression may actually be a clue to the diagnosis of FS-DFSP, while RNA in situ hybridization analysis of PDGFB expression can offer an alternative means of identifying FS-DFSP with improved sensitivity and specificity.
The diagnosis of FS-DFSP has benefited from the increasing access to and use of molecular diagnostic techniques that enable more routine detection of the recurrent PDGFB and PDGFD rearrangements that are characteristic of DFSP. Importantly, the finding of such a rearrangement helps to distinguish a tumor as DFSP, but does not separate FS-DFSP from its conventional counterpart. Instead, a growing body of literature suggests that it is a heterogeneous combination of other oncogenic alterations that coincide with FS transformation, as reflected in the increased TMB of FS-DFSP. FS transformation additionally involves loss of tumor suppressor function, although no single tumor suppressor is recurrently affected, as well as TERT promoter mutations in a minority of tumors. Thus, the available molecular genetic portrait of FS-DFSP indicates diverse secondary genetic alterations that likely are reflected in the variable phenotypes and clinical outcomes of FS-DFSP.
To better understand heterogeneity among FS-DFSP, and in doing so to effectively tailor treatment approaches to individual patients, future studies should be aimed at an integrated analysis of histology, molecular genetic pathology, and clinical outcomes. Such studies may allow for identification of subtypes of FS-DFSP with distinct biological potential that warrant more aggressive therapeutic interventions; uncovering biomarkers of these subtypes would also have tremendous clinical impact as it would allow for prospective delineation of tumors needing special treatment. Application of techniques such as DNA methylation analysis or gene expression profiling (by RNA sequencing or other methods) will allow for detailed epigenetic interrogation of tumors, going beyond DNA-level alterations to identify mechanisms of FS transformation and to translate this mechanistic insight into improved diagnostic approaches and more effective therapies. In doing so, we can hope to shed light not only on the enigmatic phenomenon of FS transformation in DFSP, but also on the process of tumor evolution more generally.
Footnotes
Ethical Considerations
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Trial Registration
Not applicable, because this article does not contain any clinical trials.