
Abstract
Diagnosing low-grade spindle cell lesions of the skin can be challenging. In particular, distinguishing among fibroblastic, myofibroblastic, and smooth muscle or pericytic proliferations may be difficult during a routine histologic examination. This article provides an update on recent developments in superficial mesenchymal tumors exhibiting smooth muscle and pericytic differentiation. We focus on the characterization of clinicopathological features, differential diagnosis, and recurrent molecular alterations. Additionally, we address superficial leiomyosarcomas, which are now classified as atypical intradermal smooth muscle neoplasms due to their low risk of aggressive behavior. The article also discusses Epstein–Barr virus-associated smooth muscle tumors.
Keywords
Introduction
Superficial tumors exhibiting smooth muscle and/or pericytic differentiation are typically suspected based on morphological features and histological architectural patterns.1–8 However, they can occasionally present diagnostic challenges, necessitating supplementary immunohistochemical studies for a definitive diagnosis.1–16 Moreover, some of these tumors are associated with Epstein–Barr virus (EBV) infections, and their biological behavior remains partially uncertain due to their rarity.2–5 Certain pericytic lesions or glomus tumors demonstrate molecular alterations that can aid in diagnosis, although these molecular changes are only required for definitive diagnosis in selected tumors where morphology and immunohistochemical findings are inconclusive.1–12 Tumors previously classified as leiomyosarcomas are now preferentially referred to as atypical intradermal smooth muscle neoplasms, given their low risk of metastasis or local recurrence.2,4–8 This reclassification applies only to intradermal tumors and to those showing limited involvement of the subcutis; however, the term cutaneous leiomyosarcoma remains acceptable under the latest World Health Organization (WHO) Classification of Skin Tumors, even for lesions with exclusively intradermal involvement.2,4–8 Recently, superficial neoplasms with GLI1 alterations have been described, which exhibit histological similarities to glomus tumors.5–9 In the present review, we aim to delineate the primary morphological, immunohistochemical, and molecular characteristics of these neoplasms with smooth muscle and pericytic differentiation, alongside their differential diagnosis with other superficial neoplasms. A comprehensive discussion of all superficial spindle cell neoplasms lies beyond the scope of this review.
Tumors With Smooth Muscle Differentiation
Superficial Benign Smooth Muscle Neoplasm
Cutaneous Leiomyomas
Clinical and Macroscopic Features
Cutaneous leiomyomas are the prototypical benign neoplasms of smooth muscle origin and are classically subdivided into pilar and genital types.4–6,13,17,18 It is usually present as a firm, skin-colored or reddish-brown nodule or papule, sometimes multiple, and can be painful, especially with cold or pressure.4–6 Both pilar and genital leiomyomas are typically small, rarely exceeding 2 cm in greatest dimension.4,17–20 Multiple pilar leiomyomas are often inherited in an autosomal dominant manner.4,16,21–24 In this context, hereditary leiomyomatosis and renal cell carcinoma syndrome is caused by heterozygous germline loss-of-function mutations in the fumarate hydratase gene, which confers susceptibility to cutaneous and uterine leiomyomas as well as an increased risk of renal cell carcinoma.4,21–24
Histopathology, Immunohistochemistry, Molecular Biology, Differential Diagnosis, and Prognosis
Pilar leiomyomas are poorly circumscribed dermal nodules (Figure 1 A, B) composed of intersecting fascicles of well-differentiated smooth muscle. The tumor cells are spindle-shaped, with elongated, blunt-ended nuclei, occasional perinuclear vacuoles, and abundant eosinophilic cytoplasm.1–6,17,18 Enlarged cells similar to those observed in symplastic uterine leiomyomas have been reported4,18 (Figure 1C and D). Nuclear enlargement and hyperchromasia may be present, and mitotic figures can be observed but are typically rare; importantly, infiltration of the subcutaneous adipose tissue is absent.1–6,17,18 The presence of mitotic figures in conjunction with cytologic pleomorphism may raise concern for atypical intradermal smooth muscle neoplasms in the differential diagnosis; this distinction is of particular importance, as cutaneous leiomyosarcoma may closely mimic leiomyoma.1–6,17,18

Cutaneous leiomyoma. (A, B) Poorly circumscribed dermal nodule composed of intersecting fascicles of well-differentiated smooth muscle (A, H&E, 100×; B, H&E, 200×). (C, D) Focally enlarged cells consistent with leiomyoma with symplastic changes (H&E, 400×). (E) Strong and diffuse membranous desmin expression (400×). (F) Strong and diffuse cytoplasmic h-caldesmon expression (200×). H&E = hematoxylin and eosin.
Immunohistochemically, markers of smooth muscle differentiation, most commonly smooth muscle actin (SMA), desmin, and h-caldesmon(Figure 1 E and F) are expressed in the vast majority of tumors.4–6,17–20 Among these, h-caldesmon appears to be the most specific marker, and the demonstration of at least 2 smooth muscle markers is generally required to support the diagnosis; various combinations of smooth muscle marker positivity may be encountered. Loss of fumarate hydratase expression (Figure 2) and increased expression of 2-succinocysteine (2SC) are useful adjuncts for identifying tumors associated with hereditary leiomyomatosis.4,20–24

Fumarate hydratase-deficient cutaneous leiomyoma associated with germline FH mutation. (A) Poorly circumscribed dermal nodule (H&E, 100×). (B) Spindle-shaped tumor cells with elongated, blunt-ended nuclei and abundant eosinophilic cytoplasm (H&E, 400×). (C) Strong and diffuse membranous h-caldesmon expression (400×). (D) Strong and diffuse smooth muscle actin expression (200×). (E) Retained cytoplasmic Succinate dehydrogenase B expression in tumor cells (200×). (F) Loss of fumarate hydratase expression in tumor cells, with positive non-tumoral cells serving as an internal positive control (200×). FH = fumarate hydratase; H&E = hematoxylin and eosin.
The diagnosis of cutaneous leiomyomas does not typically require molecular testing in sporadic tumors unless specific morphologic or immunophenotypic features raise concern for an alternative diagnosis or a syndromic association. 4 In such settings, the immunohistochemical profile may provide critical diagnostic information and, in selected tumors, prompt additional molecular investigations. In the familial setting, although fumarate hydratase and 2SC immunohistochemistry may facilitate the identification of fumarate hydratase-deficient neoplasms, they do not obviate the need for genetic testing, as biallelic fumarate hydratase mutation or inactivation may arise as a purely somatic phenomenon.4,20–24
The differential diagnosis encompasses other neoplasms with smooth muscle differentiation, including primary cutaneous (smooth muscle hamartoma (Figure 3), atypical intradermal smooth muscle neoplasms/cutaneous leiomyosarcoma and angioleiomyoma), subcutaneous leiomyosarcoma, and metastatic cutaneous lesions (uterine or soft tissue well-differentiated leiomyosarcoma), as well as neural, melanocytic (notably spindle cell and desmoplastic melanoma), myofibroblastic, fibroblastic, fibrohistiocytic tumors, and superficial NTRK-rearranged neoplasms.2,8,17–28 Many of these entities may express SMA, thereby entering the differential diagnosis. In most tumors, careful assessment of morphologic features in combination with a focused immunohistochemical panel is sufficient to resolve these diagnostic challenges.

Smooth muscle hamartoma. (A-C) Haphazardly arranged mature smooth muscle bundles in the dermis (A, H&E, 100×; B, H&E, 200×; C, H&E, 400×). (D) Diffuse smooth muscle actin expression in smooth muscle bundles (100×). (E) Diffuse desmin immunoreactivity in smooth muscle bundles (200×). (F) Strong and diffuse h-caldesmon expression in smooth muscle bundles (200×). H&E = hematoxylin and eosin.
Distinguishing cutaneous leiomyomas from smooth muscle hamartomas and atypical intradermal smooth muscle neoplasms represents one of the greatest diagnostic challenges,25–28 particularly in leiomyomas exhibiting mitotic figures.4–20 Clinical presentation is crucial for differentiating leiomyomas from smooth muscle hamartomas, as the latter are typically flat, poorly circumscribed lesions,26–28 whereas leiomyomas usually present as well-defined nodules. Regarding atypical intradermal smooth muscle neoplasms, some authors have proposed that superficial smooth muscle tumors displaying mitotic figures and cytologic atypia exceeding that expected for a typical leiomyoma should be classified as atypical intradermal smooth muscle neoplasms/cutaneous leiomyosarcoma.4–7,14–20 However, there is no clear consensus on the number of mitotic figures required to classify a tumor as an atypical intradermal smooth muscle neoplasm/cutaneous leiomyosarcoma rather than as a leiomyoma with mitotic figures. In such tumors, immunohistochemical or molecular studies generally do not provide definitive discrimination between leiomyoma and atypical intradermal smooth muscle neoplasms/cutaneous leiomyosarcoma, making careful morphologic and clinical assessment essential for an accurate diagnosis.
EBV-Associated Smooth Muscle Tumor
Clinical and Macroscopic Features
EBV-associated smooth muscle tumor is a rare, typically benign smooth muscle neoplasm that most commonly arises in the setting of immunosuppression.4–7,29–37 Superficial lesions have been reported predominantly in adult patients. Overall, these tumors are usually well-circumscribed and exhibit pink-tan to whitish cut surfaces.4,29–34
Histopathology, Immunohistochemistry, Molecular Biology, Differential Diagnosis, and Prognosis
Histologically, the majority of EBV-associated smooth muscle tumors are well-circumscribed and composed of interlacing fascicles of spindle-shaped myoid cells set within a collagenous stroma, occasionally exhibiting focal myxoid change and accompanied by focal or diffuse lymphoid infiltration (Figure 4A-D).4,29–34 Mitotic figures are generally infrequent; however, tumors with increased mitotic figures, necrosis, and nuclear pleomorphism have been reported.4,35–37

EBV-associated smooth muscle tumor. (A) Well-circumscribed subcutaneous EBV-associated smooth muscle tumor (H&E, 100×). (B) Interlacing fascicles of spindle-shaped myoid cells set within a collagenous stroma, accompanied by lymphoid infiltration (H&E, 200×). (C, D) Fascicles of spindle-shaped myoid cells with bland nuclei and focal hyperchromatic nuclei and atypia, within a collagenous stroma containing lymphocytes (H&E, 200×). (E) Strong and diffuse h-caldesmon immunoreactivity (200×).(F) In situ hybridization for Epstein–Barr virus-encoded small RNA chromogenic in situ hybridization (EBER CISH), demonstrating nuclear positivity (200×). EBV = Epstein–Barr virus; H&E = hematoxylin and eosin.
Immunohistochemically, EBV-associated smooth muscle tumor typically expresses smooth muscle markers (Figure 4E). Confirmation of EBV infection (Figure 4F) is essential for diagnosis and can be achieved either by immunohistochemistry or in situ hybridization for EBV-encoded small RNA (EBER chromogenic in situ hybridization).4,29–37
From a molecular perspective, aberrations involving the mTOR/AKT pathway and MYC overexpression have been documented; however, EBV infection in neoplastic myoid cells is most reliably demonstrated by EBER in situ hybridization.
The main differential diagnoses include other smooth muscle tumors, such as leiomyoma, angioleiomyoma, and atypical intradermal smooth muscle neoplasms/cutaneous leiomyosarcoma; Kaposi sarcoma (HHV8-positive); inflammatory myofibroblastic tumor (Anaplastic Lymphoma Kinase [ALK]-positive); and Immunoglobulin G4-related disease/inflammatory pseudotumor (IgG4-positive).2–7,13–16,29–37 Notably, none of these entities show nuclear EBER positivity, making this a crucial diagnostic feature.
EBV-associated smooth muscle tumors are generally regarded as tumors with benign clinical behavior, which appears to be more related to the patient's immune status than to histologic features.4,29–37 Nevertheless, in tumors exhibiting a high mitotic rate, necrosis, or significant cytologic atypia, the clinical course remains incompletely understood. Given the limited number of reported tumors, larger series with extended follow-up are needed to determine whether EBV-associated smooth muscle tumors consistently pursue a benign clinical course.
Superficial Intermediate Smooth Muscle Neoplasm
Atypical Intradermal Smooth Muscle Neoplasms/Cutaneous Leiomyosarcoma
Clinical and Macroscopic Features
Cutaneous leiomyosarcoma accounts for 2% to 3% of cutaneous sarcomas (∼0.04% of all skin tumors) and is the 4th most common subtype after dermatofibrosarcoma protuberans, pleomorphic dermal sarcoma, and Kaposi sarcoma.4–7,15,38–73 Superficial leiomyosarcoma is classified as cutaneous (intradermal), subcutaneous, or metastatic, with prognosis closely related to tumor depth. 14
The introduction of the concept of “atypical intradermal smooth muscle neoplasm” has led to some confusion among “superficial leiomyosarcoma,” atypical intradermal smooth muscle neoplasms, subcutaneous leiomyosarcoma, and other variants of leiomyosarcoma. Recently, Bressler et al, in an informative study of 39 subcutaneous leiomyosarcomas, reported that among the 36 tumors with available clinical data and follow-up, 12 (33%) developed metastases, with the lung representing the most common site. One patient developed local recurrence, and 6 of the 36 patients died of the disease. 74 These findings support that subcutaneous leiomyosarcomas exhibit more aggressive behavior than dermal-based tumors and have a clinical course comparable to that of deep-seated leiomyosarcomas.74,75
Dermal leiomyosarcoma is considered intermediate malignancy, with a local recurrence of ∼24% and very low metastatic potential (∼4%).76,77 Lesions confined to the dermis have not shown metastases, though limited data warrant cautious interpretation.4–7,42–45,47 A 3-tier classification reflects prognosis: (1) pure dermal leiomyosarcoma/atypical intradermal smooth muscle neoplasms) (most favorable); (2) dermal leiomyosarcoma with subcutaneous extension (intermediate); and (3) pure subcutaneous leiomyosarcoma (poorest).47,74
Diagnosis requires exclusion of metastatic leiomyosarcoma from deep or visceral sites, as cutaneous metastases indicate advanced disease (median survival ∼16 months). 43 Most superficial leiomyosarcomas arise de novo, though rare associations include prior radiotherapy, trauma, or hereditary syndromes such as Reed syndrome, Birt–Hogg–Dubé, and Li–Fraumeni.4,44,67,68 Subcutaneous leiomyosarcoma typically affects extremities (50%-85%), especially lower limb extensors, while cutaneous leiomyosarcoma favors the scalp and face. Lesions are usually firm nodules or plaques (Figure 5A and B), slow-growing, 0.5 to 3.5 cm (up to 19 cm), with subcutaneous tumors appearing larger and more circumscribed. 70

Atypical intradermal smooth muscle neoplasm/cutaneous leiomyosarcoma. (A) Well-circumscribed cutaneous lesion of the pubic region. (B) Magnetic resonance imaging demonstrating a superficial cutaneous lesion. (C) Dermal tumor composed of interlacing fascicles of smooth muscle fibers (H&E, 100×). (D) Neoplastic cells are spindle-shaped, with elongated, blunt-ended nuclei and eosinophilic, fibrillar cytoplasm (H&E, 200×). (E) Atypical intradermal smooth muscle neoplasm/cutaneous leiomyosarcoma showing 2 distinct areas: 1 with bland cytologic features and the other with hyperchromatic nuclei (H&E, 200×). (F) Area with hyperchromatic nuclei and marked nuclear atypia, including anisokaryosis (H&E, 400×). H&E = hematoxylin and eosin.
Histopathology, Immunohistochemistry, Molecular Biology, Differential Diagnosis, and Prognosis
Two histologic growth patterns of cutaneous leiomyosarcoma have been described: nodular and diffuse. Nodular leiomyosarcoma is usually highly cellular, with nuclear atypia and mitotic figures, whereas diffuse leiomyosarcoma exhibits lower cellularity, mild pleomorphism, and fewer mitotic figures, often corresponding to poorly circumscribed dermal proliferations that may extend into subcutaneous fat. Speculations regarding the likely histogenetic origin—namely, arrector pili muscles versus vascular smooth muscle cells, have been proposed. The histological distribution may support a possible origin; however, in some instances, particularly in tumors with a diffuse growth pattern, it is difficult to assign a definitive lineage. Moreover, immunohistochemical findings do not reliably support a specific origin. Notably, derivation from either arrector pili muscle or vascular smooth muscle cells does not appear to confer a distinct prognostic implication or influence tumor behavior.48,49 Epidermal changes such as ulceration, acanthosis, or effacement of rete ridges may also be observed. 72
Subcutaneous leiomyosarcoma is generally well-circumscribed, compresses adjacent structures, and is confined to the subcutis, sparing the dermis. Both cutaneous and subcutaneous leiomyosarcoma display interlacing fascicles of spindle cells with elongated, blunt-ended nuclei, eosinophilic fibrillar cytoplasm (Figure 5C-F), mitotic figures (Figure 6A), and frequently a perinuclear clear halo.4–7,14–16,38–62,76,77 Histologic variants include epithelioid, granular cell, sclerotic, pleomorphic, and multinucleated giant cell leiomyosarcoma, sometimes with prominent desmoplastic or myxoid stroma.4–7,14–16,39,50

Atypical intradermal smooth muscle neoplasm/cutaneous leiomyosarcoma. (A) Spindle-shaped neoplastic cells with elongated, blunt-ended nuclei, inconspicuous nucleoli, nuclear hyperchromasia, and mitoses (H&E, 400×). (B) Strong and diffuse h-caldesmon expression in tumor cells (400×). H&E = hematoxylin and eosin.
Immunohistochemistry is essential to distinguish leiomyosarcoma from other spindle cell tumors. Well-differentiated leiomyosarcoma expresses desmin, h-caldesmon (Figure 6B), SMA, muscle-specific actin, and myosin.4–7,14–16,42 Poorly differentiated or subcutaneous lesions may lose desmin.4–7 At least 2 smooth muscle markers are recommended for diagnosis. Leiomyosarcoma is typically S100-negative, and other markers (CD34, CD31, Epithelial membrane antigen, keratins, -Human Melanoma Black-45 [HMB45], Melanoma Antigen [MELAN A], SRY-related HMG-box/SOX10, Microphthalmia-associated transcription factor) help exclude spindle cell melanoma, spindle cell carcinoma, or vascular tumors.4–7,14,17,42,70 Superficial malignant peripheral nerve sheath tumor is rare; when this diagnosis is suspected, the possibility of desmoplastic or spindle cell melanoma should always be excluded. In some leiomyosarcomas, distinction from primary dermal sarcoma may be challenging, particularly when the expression of lineage markers (ie, smooth muscle differentiation markers) is focal or equivocal.
Cellular atypia and ≥1 mitotic figures per 10 high power fields suggest leiomyosarcoma (Figure 6A); however, occasional mitotic figures can also be observed in leiomyomas, 4 complicating the distinction between well-differentiated leiomyosarcoma and leiomyomas with mitotic figures. Ki-67 and Phosphohistone H3 can aid in challenging tumors, but generally do not provide conclusive information for a definitive diagnosis. 47
Margin status is the main predictor of recurrence. Given the low metastatic risk of dermal leiomyosarcoma, these tumors have been renamed atypical intradermal smooth muscle neoplasms, although the term cutaneous leiomyosarcoma remains acceptable under the latest WHO classification.4–7,42 Atypical intradermal smooth muscle neoplasms show increased cellularity, nuclear enlargement, hyperchromatism, and occasional mitotic figures but rarely exhibits marked pleomorphism, high mitotic index, or necrosis.4–7,42 They are generally indolent, with excellent prognosis after complete excision; metastasis has not been documented,4–7,42 and tumor grading does not predict outcome.38,73
Molecular data on cutaneous leiomyosarcoma, mainly confined to the dermis or with minimal subcutaneous involvement, are limited. 63 Recurrent TP53 and RB1 mutations, ultraviolet-related mutational signatures (SBS7a/b, DBS1), somatic copy number alterations (deletions: TP53, KDM6B, CYLD; amplifications: ZMYM2, MYOCD, MAP2K4, NCOR1), and gene fusions (CRTC1/CRTC3::MAML2) have been reported. 63 Atypical intradermal smooth muscle neoplasm-like lesions in hereditary leiomyomatosis show fumarate hydratase germline mutations and 2-succinyl-cysteine expression.4–7,44,78
Recently, Alston et al described a series of pediatric soft tissue tumors with smooth muscle differentiation and identified 2 apparently distinct clinical groups. 79 Group 1 tumors are composed of mildly to moderately atypical ovoid to spindle cells with eosinophilic cytoplasm; they show a perivascular growth pattern, exhibit mitotic figures, lack necrosis, and some tumors harbor SRF rearrangements. In contrast, Group 2 tumors are highly cellular and pleomorphic, with brisk mitotic figures and biallelic TP53 inactivation. 79 The prognosis for Group 1 tumors is excellent, and based on these findings, the term “pediatric-type myoid neoplasms of somatic soft tissue” has been proposed for Group 1, whereas the designation of true leiomyosarcoma is restricted to Group 2. 79 This study has limited applicability, as the majority of the leiomyosarcomas analyzed were deep-seated, and the article does not specify whether the superficial tumors were subcutaneous or dermal in origin. 79 Consequently, the applicability of this classification to cutaneous smooth muscle tumors remains uncertain.
Benign Pericytic and Perivascular Neoplasms
Angioleiomyoma
Clinical and Macroscopic Features
Angioleiomyoma is a benign mesenchymal tumor that frequently arises in the dermis or subcutis. It consists of thick-walled vessels surrounded by well-differentiated perivascular smooth muscle bundles and belongs to the perivascular myoid tumor family, forming a morphological continuum with myopericytoma.4–7,79–82 These tumors most commonly occur in the skin and subcutis (Figure 7A) of the extremities, especially around the knee.4–7,79–88 The lesions are usually solitary, small (<30 mm), firm, well-circumscribed (Figure 7A), and slow-growing nodules, with roughly half being painful.2–7,79–88

Cutaneous angioleiomyoma. (A) Well-circumscribed subcutaneous nodule (H&E, 100×). (B-D) Well-differentiated smooth muscle surrounding blood vessels of variable caliber (B, H&E, 200×; C, D, H&E, 400×). (E) Strong and diffuse membranous smooth muscle actin expression (200×). (F) Strong and diffuse h-caldesmon immunoreactivity (200×). H&E = hematoxylin and eosin.
Histopathology, Immunohistochemistry, Molecular Biology, Differential Diagnosis, and Prognosis
Angioleiomyomas are benign tumors composed of bland, well-differentiated smooth muscle surrounding blood vessels of variable caliber,1–7,79–88 with inconspicuous mitotic figures (Figure 7B-D). Three vascular subtypes are recognized—solid, venous, and cavernous—and hybrid forms may occur; occasionally, some neoplasms may show myopericytoma-like concentric perivascular whorling.4–7,80–85 Tumor cells are diffusely positive for SMA, calponin, and h-caldesmon (Figure 7E and F) with variable or absent desmin.4–7,80–85 Almost all so-called “angiomyolipomas” in the skin represent angioleiomyomas with adipocytic metaplasia and should not be confused with true angiomyolipomas associated with tuberous sclerosis complex. As expected, HMB45 is consistently negative in this tumor.82,83 Cytogenetic alterations include monosomy 13 and losses of 6p, 21q, and 13q, with rare NOTCH gene fusions and no recurrent PDGFRB mutations.4–7,9,10,84,85
Angioleiomyoma can be confused with several other lesions. Leiomyoma exhibiting collapsed vascular structures secondary to extensive collagenization may present a diagnostic challenge, as it can closely mimic angioleiomyoma. Glomus tumors are a frequent mimic because it is also vascular and often painful; however, glomus tumors are typically smaller, arise in subungual locations or the distal extremities, and are composed of round, uniform cells arranged around vessels rather than spindle-shaped smooth muscle cells.1–7,80–87 Myopericytoma may also resemble angioleiomyoma, as both are perivascular neoplasms; however, myopericytoma characteristically shows concentric layers of spindle myoid cells surrounding vessels and most commonly occurs in the upper extremities. Hemangioma or spindle cell hemangioma may appear similar grossly, but these lesions are primarily vascular and lack prominent smooth muscle bundles. Rarely, solitary fibrous tumors or low-grade sarcomas may enter the differential diagnosis; however, these typically demonstrate cellular atypia, increased mitotic figures, or a more fibrous growth pattern, along with STAT6 positivity in solitary fibrous tumors.9,10,13 Angioleiomyoma is a benign tumor that rarely recurs after surgical excision.
Myofibroma and Myofibromatosis
Clinical and Macroscopic Features
Myofibromas are benign mesenchymal neoplasms of presumed myofibroblastic origin. They occur in 3 clinicopathologic forms: solitary, multicentric, and generalized.1–7,89–93 The solitary form shows a male predominance, whereas multicentric disease predominantly affects females. Clinically, myofibromas are present as well-circumscribed but unencapsulated, firm, violaceous dermal or subcutaneous nodules ranging from a few millimeters to several centimeters in size.89–93 Solitary lesions most often arise in the skin or subcutis of the head and neck, upper extremities, or trunk, with a similar distribution in multicentric disease. Myofibroma accounts for approximately 12% of pediatric soft tissue tumors; 90% of tumors occur in children. Multicentric lesions are exclusively pediatric.4,90–93
Histopathology, Immunohistochemistry, Molecular Biology, Differential Diagnosis, and Prognosis
Myofibroma shows a characteristic biphasic growth pattern, with alternating areas of primitive, darkly staining small round cells and plump spindle-shaped mesenchymal cells, accompanied by numerous thin-walled hemangiopericytoma-like vessels and myointimal-type nodular proliferations (“vascular balls”)4–7,86,89–95 (Figure 8). These features support a relationship to other perivascular myoid neoplasms and account for their morphological overlap.4,86,89–95 Mitotic figures are usually infrequent but may be increased, and focal ischemic-type coagulative necrosis may be present. 4 Tumor cells are diffusely positive for SMA, while h-caldesmon is absent or only focally expressed; desmin is typically negative.4–7,86,89–91

Myofibroma. (A-C) Myofibroma at low power showing a multinodular subcutaneous lesion (H&E, 40×). (B, C) Lesion demonstrates a biphasic growth pattern with myointimal-type nodular proliferations (“vascular balls”) (B, H&E, 200×; C, H&E, 400×). (D) Myofibroma with multinodular growth pattern in a subcutaneous location (H&E, 100×). (E, F) Perivascular myoid neoplasms with myointimal-type nodular proliferations and areas resembling nodular fasciitis (E, H&E, 200×; F, H&E, 400×). H&E = hematoxylin and eosin.
Familial tumors harbor germline PDGFRB (≈89%) or NOTCH3 (≈11%) mutations,86,93–96 whereas sporadic solitary lesions more frequently exhibit somatic PDGFRB mutations, 93 with distinct mutational spectra observed between familial and sporadic disease. SRF rearrangements have been reported in highly cellular tumors.4,86,95
Myofibroma can resemble a variety of other tumors and lesions. Other benign myofibroblastic tumors, such as fibromatosis or nodular fasciitis, may appear similar; however, fibromatosis is typically deeper and more infiltrative, whereas nodular fasciitis often shows rapid growth and a more myxoid stroma and is associated with Ubiquitin Specific Peptidase 6 rearrangements.4,93 Smooth muscle tumors, including leiomyomas, may also resemble myofibroma histologically, but they are composed of more uniform spindle cells and show desmin positivity. Solitary fibrous tumor can closely mimic myofibroma because of its characteristic staghorn vascular pattern; however, immunohistochemistry (eg, CD34 and STAT6) is useful in distinguishing these entities. 13 Infantile hemangiomas are more purely vascular and lack a myofibroblastic component. Rarely, malignant neoplasms must be considered in the differential diagnosis. Leiomyosarcoma, fibrosarcoma, and embryonal rhabdomyosarcoma may mimic myofibroma but typically demonstrate nuclear atypia, increased mitotic figures, necrosis, or evidence of skeletal muscle differentiation. Spindle cell neural tumors, such as schwannoma or neurofibroma, may also enter the differential diagnosis and usually show S100 positivity. Inflammatory myofibroblastic tumors can appear similar because they contain myofibroblasts admixed with inflammatory cells, and a subset, particularly in children, shows ALK positivity.4–7
Myofibroma is a benign neoplasm and is typically cured by complete surgical excision; however, the generalized form is often fatal due to extensive visceral involvement and limited surgical options.
Myopericytoma
Clinical and Macroscopic Features
Myopericytoma is a benign neoplasm with perivascular myoid differentiation. It most commonly involves the distal extremities and presents as painless, dermal or subcutaneous nodules.1–7,96–100 The tumor occurs across a wide age range and myopericytoma is thought to differentiate toward myopericytes.96–100 Clinically, lesions are well-circumscribed, firm, superficial nodules, typically measuring less than 20 mm in diameter.96–100
Histopathology, Immunohistochemistry, Molecular Biology, Differential Diagnosis, and Prognosis
Myopericytoma is a well-circumscribed, unencapsulated nodule composed of cytologically bland oval-to-spindle cells arranged in concentric, multilayered perivascular patterns,1–7,96–102 with eosinophilic myoid cytoplasm and round to ovoid nuclei (Figures 9 and 10). Cellularity and vascular architecture are variable; thin-walled communicating vessels may mimic solitary fibrous tumor, thicker vessels resemble angioleiomyoma (Figure 9D-F), and solid cellular areas overlap with myofibroma (Figure 10). Rare variants include diffuse plexiform growth (myopericytomatosis) and intramural and intravascular forms. Uncommon features include hyalinization, pseudopapillary vascular architecture (Figure 10A-D), metaplastic bone, ischemic necrosis, and focal myxoid stromal tissue (Figure 10F). Tumor cells are typically positive for SMA and h-caldesmon, and usually negative or only focally positive for desmin.1–7,96–102 Although historically grouped with myofibroma, angioleiomyoma, and glomus tumor, recent molecular data support these as distinct entities despite overlapping morphology and phenotype.1–7,96–102 The differential diagnosis includes myofibroma, angioleiomyoma, and solitary fibrous tumor.

Myopericytoma. (A) Well-circumscribed subcutaneous myopericytoma with lymphoid infiltration (H&E, 100×). (B, C) Myopericytoma composed of oval-to-spindle-shaped cells arranged in concentric, multilayered perivascular patterns, with eosinophilic myoid cytoplasm, ovoid nuclei, and lymphoid infiltration (H&E, 400×). (D) Well-circumscribed subcutaneous myopericytoma with collagen deposition (H&E, 100×). (E, F) Myopericytoma with tumor cells arranged in concentric, multilayered perivascular patterns and sparse lymphoid infiltration (E, H&E, 200×; F, H&E, 400×). H&E = hematoxylin and eosin.

Hypercellular myopericytoma with focal pseudopapillary formation. (A) Well-circumscribed subcutaneous myopericytoma with hypercellularity and focal pseudopapillary architecture (H&E, 100×). (B, C) Myopericytoma with bland neoplastic cells arranged in concentric, multilayered perivascular patterns and focal lymphoid infiltration (B, H&E, 200×; C, H&E, 400×). (D) Pseudopapillary formation in myopericytoma (H&E, 100×). (E) Hypercellular myopericytoma with small vessels and concentric tumor cell growth (H&E, 200×). (F) Myopericytoma with focal myxoid stromal tissue and sparse lymphoid infiltration (H&E, 400×). H&E = hematoxylin and eosin.
Myopericytoma can be confused with myofibroma, which is more common in children and has a biphasic pattern with hemangiopericytoma-like vessels, whereas myopericytoma has concentric spindle cells around vessels. Glomus tumors can look similar, especially around small blood vessels, but they are usually painful, small, and have round, uniform cells rather than spindle cells.101–105 Angioleiomyomas are smooth muscle tumors around vessels that are more fascicular and often desmin-positive, unlike myopericytoma, which has a concentric pattern. Solitary fibrous tumors can have similar vascular patterns, but they are usually CD34 and STAT6-positive and lack the concentric arrangement of myopericytoma. 13 Rarely, other perivascular tumors or low-grade sarcomas may mimic it, but these usually show atypia, mitotic figures, or necrosis. Immunostaining helps confirm myopericytoma, as the tumor cells are usually SMA, desmin variable, and CD34-negative. Approximately 15% of myopericytomas harbor BRAF mutations, though data remain limited.4–7,108 Reported PDGFRB mutation data in myopericytoma are inconsistent.99,106–108 Myopericytoma is a benign lesion with no reported recurrences or metastases.
Glomus Tumors
Clinical and Macroscopic Features
Glomus tumors are benign mesenchymal neoplasms of modified glomus body cells, most common in adults (4th-6th decades) and rarely in children.4–7,102–105,109–116 They usually present as small (<10 mm), painful, firm, solitary nodules, or plaques in distal extremities, especially subungual regions, with pink-to-blue coloration.4–7,101–105,109–111 Rare malignant glomus tumors have been reported.102,103,105,110,112–114
Histopathology, Immunohistochemical, Molecular Biology, Differential Diagnosis, and Prognosis
Glomus cells are small, uniform, round cells with centrally located nuclei, amphophilic to lightly eosinophilic cytoplasm, sharply defined borders, and a peripheral basal lamina (Figure 11A-C).4–7,85,101–105,109–113,115–117 Recognized histologic patterns include oncocytic, epithelioid, myxoid, and hyalinized variants.4–7,85,102–105,109–113,115–117 Solid glomus tumors account for ∼75% of lesions and consist of nests of glomus cells surrounding capillary-sized vessels; small cuffs of glomus cells often occur around adjacent vessels outside the main tumor. Glomangiomyoma, glomangiopericytoma, and symplastic glomus are alternative variants.4–7,101,104,105,109–111

Glomus tumor. (A) Well-circumscribed dermal glomus tumor (H&E, 100×). (B, C) Sheets of densely packed cells with small, round, uniform, centrally located nuclei and scant amphophilic to lightly eosinophilic cytoplasm, with a peripheral basal lamina (H&E, 200×). (D) Dendritic cells show S100 positivity, while glomus tumor cells are S100-negative (100×). (E) Strong and diffuse smooth muscle actin immunoreactivity (200×). (F) Diffuse and membranous collagen IV immunoreactivity in tumor cells (400×). H&E = hematoxylin and eosin.
Malignant glomus tumors are extremely infrequent.102–105109 Diagnosis requires marked nuclear atypia (with any mitotic figures) and/or atypical mitotic figures, and malignant tumors may retain conventional architecture or show spindled areas resembling leiomyosarcoma or fibrosarcoma.4–7,85,101–105,109–120 In the absence of a benign precursor, immunohistochemistry for SMA and pericellular collagen IV is needed to confirm round cell malignant glomus tumors.4–7,85,101–105,109–120
Glomus tumors consistently express SMA, h-caldesmon, and pericellular collagen IV (Figure 11D-F).4–7,85,102–105,109–120 S100 is typically negative in glomus tumors; however, dendritic cells in the stromal tissue show S100 expression (Figure 11D). Detection of MIR143::NOTCH1/2/3 fusions can aid diagnosis in selected tumors; sporadic glomus tumors frequently harbor rearrangements involving MIR143 and NOTCH genes.4,117 BRAF p.V600E mutations occur in a small subset of histologically malignant tumors.4,115
Glomus tumors can be confused with several other lesions. Subungual melanoma may mimic a glomus tumor, especially if pigmented, but melanoma usually has irregular pigmentation, rapid growth, and cytologic atypia.4–7,85,101–105,109–111 Mucoid cysts or digital myxoid cysts may appear as small distal finger nodules but are usually painless, soft, and transilluminate. Hemangioma or vascular malformations can mimic glomus tumors visually, but hemangiomas are often larger, more superficial, and less painful, and histologically show endothelial proliferation rather than glomus cells. Neuroma or neurofibroma may cause pain but are typically firm, less tender to cold, and S100-positive, unlike glomus tumor, which is SMA-positive.4–7,102–105,109–111 Epidermal inclusion cysts can occur in the fingertip but are usually painless unless infected. Rarely, angioleiomyoma or myopericytoma may mimic glomus tumors, but these are deeper, spindle-cell perivascular tumors, whereas glomus tumors have small, round, uniform cells clustered around vascular channels. Immunohistochemistry shows SMA-positive, desmin variable, S100-negative, helping to distinguish it from neural or fibrohistiocytic lesions.4–7
GLI1-altered tumors represent an emerging differential diagnosis.122–125,126,127 These neoplasms may occur superficially and often display a glomoid architecture, composed of monotonous epithelioid cells that are round to ovoid, with occasional spindle or pleomorphic areas. 122–125,126,127 The stroma is scant and collagenous or focally myxoid. Additional features include a perivascular growth pattern with lymphovascular invasion or protrusion (Figure 12B) and variable mitotic figures (Figure 12C). 122–125,126,127 Immunohistochemically, tumor cells are positive for GLI1 (Figure 12D), 122–125,126,127 with variable expression of S100 and CD56. GLI1-amplified tumors may also show variable immunoreactivity for murine doble minute 2 (MDM2) (Figure 12E), STAT6, Damage Inducible Transcript 3 (DDIT3), CDK4, and p16, depending on the size of the amplicon and associated coamplifications. 122–125,126,127 In contrast to conventional glomus tumors, these neoplasms generally lack expression of SMA and collagen IV. Fluorescence in situ hybridization analysis confirms GLI1 amplification (Figure 12F), frequently with coamplification of CDK4 (Figure 12G), MDM2, DDIT3, or STAT6.122–125,126,127 DDIT3 fluorescence in situ hybridization showing a positive translocation may also raise concern for a possible GLI1 rearrangement 127 ; however, RNA sequencing usually provides critical information regarding the specific fusion partner.

Superficial GLI1-amplified tumor. (A) Cutaneous recurrence of a GLI1-amplified tumor (macroscopic photograph courtesy of Dr Celia Requena, IVO). (B) Poorly circumscribed dermal and subcutaneous tumor with sheets of densely packed cells and lymphovascular invasion/protrusion of tumor cells into endothelial-lined vascular spaces (H&E, 200×). (C) Sheets of uniform tumor cells with small, round-to-epithelioid morphology, bland nuclei with small nucleoli, and mitotic figures; small blood vessels in the background (H&E, 400×). (D) Diffuse GLI1 immunoreactivity in tumor cells (200×). (E) Focal nuclear MDM2 expression in tumor cells (200×). (F) Fluorescence in situ hybridization (FISH) showing GLI1 amplification (clusters of red signals) (600×). (G) FISH showing coamplification of CDK4 in tumor cells (600×). H&E = hematoxylin and eosin.
Typical glomus tumors, glomuvenous malformations, and symplastic variants are benign, but malignant glomus tumors are aggressive, with a potential risk of metastases. 4,103–121
Diagnostic Challenges in Superficial Tumors With Smooth Muscle and Pericytic Differentiation
Careful evaluation of the clinical context, including anatomic site, patient age, and previous clinical history, is crucial for accurate differential diagnosis. For example, smooth muscle hamartomas and cutaneous leiomyomas with diffuse growth patterns may display similar morphology, but they are clinically distinct.
When encountering multiple cutaneous leiomyomas, the possibility of fumarate hydratase-deficient neoplasms should be considered. We recommend testing for fumarate hydratase and 2SC. Loss of fumarate hydratase expression accompanied by diffuse 2SC positivity may indicate a fumarate hydratase-deficient tumor, warranting referral for genetic counseling to rule out hereditary syndromes.
Distinguishing between leiomyomas with mitotic figures and atypical intradermal smooth muscle neoplasms/cutaneous can be challenging. There is no universally accepted mitotic threshold to classify a tumor as a superficial leiomyoma with mitotic figures versus an atypical intradermal smooth muscle neoplasm/cutaneous leiomyosarcoma. Generally, atypical intradermal smooth muscle neoplasms/cutaneous leiomyosarcoma are more cellular, show less collagen, have a diffuse infiltrative growth pattern, and display greater nuclear atypia, though exceptions exist. In practice, we prefer to classify a mitotically active neoplasm as an atypical intradermal smooth muscle neoplasm/cutaneous leiomyosarcoma; if surgical margins are positive, re-resection is advised. The risk of recurrence is very low, and in the rare event of metastasis, the tumor will already have been appropriately classified.
Subcutaneous leiomyosarcoma represents a rare and not fully characterized entity. The available literature remains limited, consisting predominantly of reports on isolated patients and small clinicopathologic series that are frequently confounded by the inclusion of both its more indolent counterpart, atypical intradermal smooth muscle neoplasm/cutaneous leiomyosarcoma, and highly aggressive deep-seated leiomyosarcomas. Subcutaneous leiomyosarcomas demonstrate more aggressive clinical behavior than dermal-based tumors and exhibit a biological course more akin to that of deep-seated leiomyosarcomas.
Superficial tumor with high histological grade, necrosis, pleomorphism tumor with smooth muscle differentiation should raise strong consideration for uterine/soft tissue or visceral metastatic leiomyosarcoma rather than atypical intradermal smooth muscle neoplasms/cutaneous leiomyosarcoma.
The classification of EBV-associated smooth muscle neoplasms with mitoses and necrosis as true sarcomas remains uncertain. EBV testing (immunohistochemical stain, chromogenic in situ hybridization, or molecular) is recommended for all superficial smooth muscle tumors arising in patients with a history of immunosuppression, in pediatric patients, or in tumors showing unusual histologic features.
Some superficial cutaneous lesions may show overlapping smooth muscle and pericytic differentiation. For example, angioleiomyomas can mimic classic pericytic lesions such as myofibroma or myopericytoma. In tumors with overlapping features, given that both entities are benign lesions, a descriptive designation such as “benign pericytic/perivascular tumor” may be appropriate.
Immunohistochemistry can support smooth muscle differentiation, while molecular testing is not mandatory for classification. However, in ambiguous or atypical tumors, molecular results may assist in reaching a final diagnosis. Molecular findings do not currently carry prognostic significance. Morphologic assessment remains the cornerstone of diagnosis. Pathologists should carefully evaluate architectural, cytologic, and stromal features to guide differential diagnosis. Although morphologic overlap is considerable, early recognition of key histologic characteristics can focus immunohistochemical workup.
Emerging entities, such as superficial GLI1-altered neoplasms, should be considered in tumors with a glomoid appearance when immunohistochemistry does not support a classic glomus tumor. The prevalence of atypical glomus tumors that may actually represent underdiagnosed GLI1-altered neoplasms remains unclear.
Conclusions
Superficial cutaneous tumors with smooth muscle and pericytic differentiation often show overlapping morphologic features and immunoprofiles, making diagnosis challenging. A systematic approach that integrates histopathologic evaluation, immunohistochemistry, and, when necessary, molecular testing improves diagnostic accuracy; however, molecular analyses are usually not required for the diagnosis of these lesions. Table 1 summarizes the main clinicopathologic features of smooth muscle and pericytic neoplasms. Some smooth muscle neoplasms have genetic implications, underscoring the importance of accurate classification for appropriate genetic counseling. Prognosis is generally favorable; nevertheless, worrisome histologic features should be documented, as rare lesions may exhibit locally aggressive behavior or metastasize. Pathologists should also be aware of newly recognized entities, such as superficial GLI1-altered neoplasms, which may mimic pericytic tumors (eg, glomus tumors).
Differential diagnosis
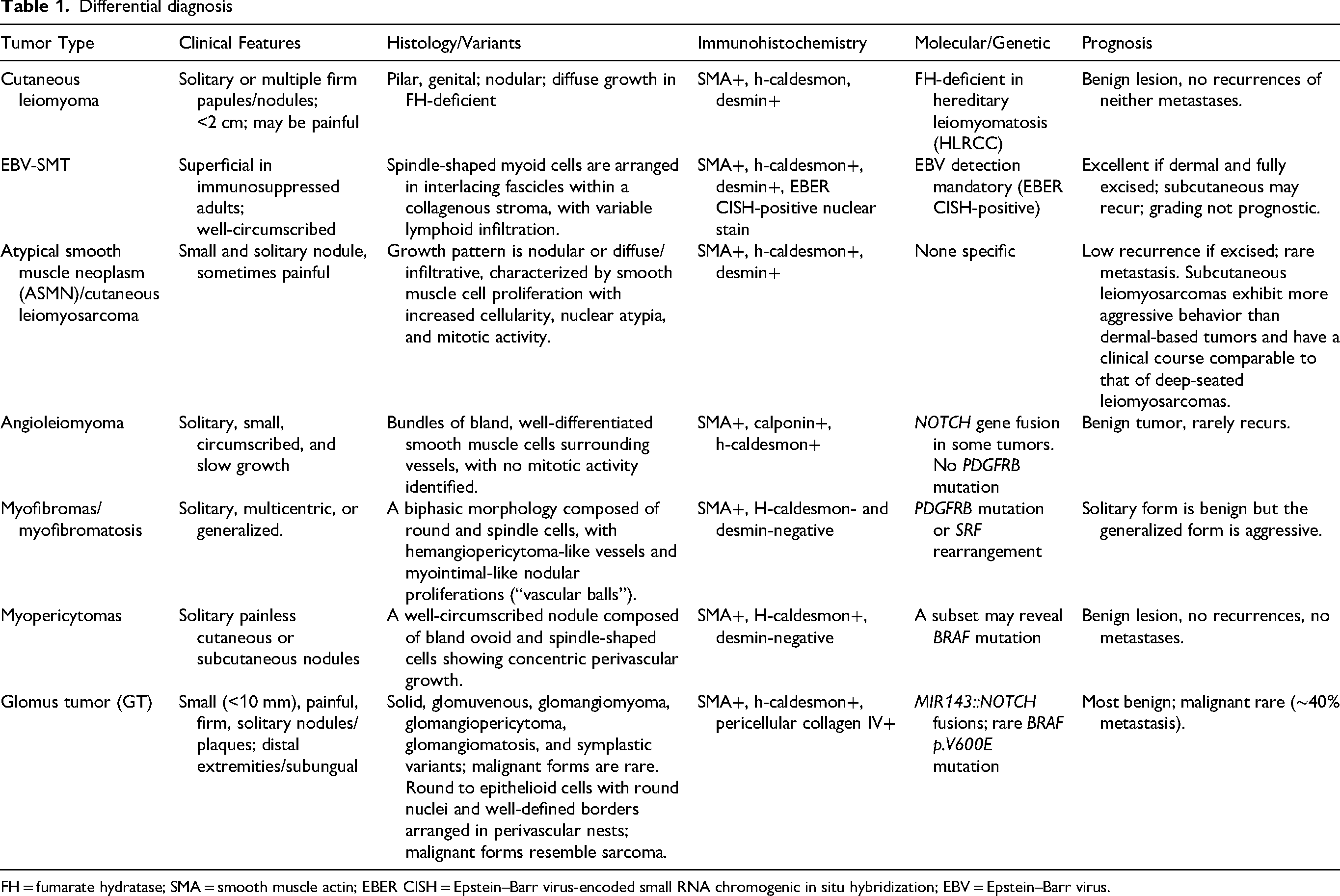
FH = fumarate hydratase; SMA = smooth muscle actin; EBER CISH = Epstein–Barr virus-encoded small RNA chromogenic in situ hybridization; EBV = Epstein–Barr virus.
Footnotes
Ethical Considerations
Ethical approval is not applicable to a review-type manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data availability is not applicable to a review-type manuscript.