
Abstract
Round cell sarcoma with EWSR1::PATZ1 fusion is an extremely rare sarcoma of soft tissue and bones that comes under EWSR1::non-ETS fusion sarcoma. Molecular studies, such as next-generation sequencing, are essential for accurate diagnosis, as this tumor presents as conventional round cell sarcoma, often co-expressing myogenic and neurogenic markers, which can prompt an erroneous diagnosis of rhabdomyosarcoma (RMS) or malignant peripheral nerve sheath tumor. Due to the rare incidence and paucity of literature on this tumor, the definite prognostic implications and therapeutic guidelines are lacking. Here, we describe two patients with EWSR1::PATZ1 sarcoma, of which one patient was initially misdiagnosed as synovial sarcoma and RMS on two occasions. These patients underscore the diagnostic challenges and therapeutic uncertainties surrounding EWSR1::PATZ1 fusion sarcomas, emphasizing the need for further large collaborative studies to establish optimal prognostic implications and management strategies for this rare entity.
Introduction
The fifth edition WHO classification of bone and soft tissue tumors includes a category of “undifferentiated round cell sarcomas (URCS) of bone and soft tissue”. This further includes, apart from Ewing sarcoma (ES), three distinct entities: CIC rearranged sarcomas, sarcomas with BCOR genetic alterations, and round cell sarcomas with EWSR1::non-ETS fusions. 1 EWSR1::non-ETS fusions comprises of EWSR1::NFATC2 sarcomas and EWSR1::PATZ1 sarcomas. EWSR1::PATZ1 is a rare fusion where an intrachromosomal rearrangement between EWSR1 and PATZ1 leads to an in-frame fusion of the N-terminal domain of EWSR1 and the zinc finger domain of PATZ1.1,2 Unlike ES, EWSR1::PATZ1 fusion sarcomas have a heterogenous presentation, presenting at a median age of around 40 years, with characteristic involvement of soft tissues of the abdomen, head and neck, and extremities. Histologically, it is characterized by diffuse small round, or spindled cells in a fibrous and myxoid stroma. On immunohistochemistry (IHC), there may be a co-expression of myogenic and neurogenic markers, which can prompt an erroneous diagnosis of rhabdomyosarcoma (RMS) or malignant peripheral nerve sheath tumor (MPNST). Molecular studies, such as high-throughput next-generation sequencing (NGS), are essential to establish the evidence-based diagnosis.3–7
These tumors are treated with conventional regime for ES; however, they are associated with an unfavorable clinical course.8,9 Due to the rarity of this entity and limited prevailing literature, there are no definite guidelines for the management of EWSR1::PATZ1 soft tissue sarcomas.5,8,9 We hereby describe two patients with EWSR1::PATZ1 fusion sarcoma, proven on molecular studies, that presented to us at a dedicated sarcoma clinic in North India.
Patient Presentation
Patient 1
A 45-year-old man presented with a lump in the right upper quadrant of the abdomen for the past 1 year, which had gradually increased in size over the last few months. He was initially evaluated elsewhere. A contrast-enhanced computed tomography (CECT) scan of the abdomen revealed a heterogeneously enhancing round-to-oval lesion in the subcutaneous plane in the right upper quadrant, measuring 3.7 × 3.4 × 3.1 cm and abutting the right rectus abdominis posteriorly, with focal loss of fat planes. Magnetic resonance imaging (MRI) findings were in resonance with the CT scan findings. A positron emission tomography (PET)-CT scan did not show any evidence of distant metastatic disease.
Trucut biopsy from the lesion was reported as URCS. He underwent wide local excision, the histopathology of which revealed an oval-to-spindled cell tumor arranged in sheets, with IHC positive for S100 and NSE, suspicious of ES and MPNST. The surgical margins were free. The patient was then referred to our tertiary cancer care center and biopsy blocks were reviewed. The tumor showed features of a malignant round-to-spindled cell tumor (Figure 1A, B), immunopositive for S100 (focal, Figure 1C), EMA, desmin, MYOD1 (focal, Figure 1D), keratin (focal); while immunonegative for NKX2.2, BCL2, myogenin and retained H3k27me3 expression. Break-apart fluorescence in situ hybridization (BA-FISH) for EWSR1, CIC and BCOR did not reveal any rearrangement. Due to the atypical site and age of presentation, NGS was performed which revealed EWSR1::PATZ1 fusion, with exon 8 of the EWSR1 gene at the 5′ end being fused with exon 1 of PATZ1 gene at the 3′ end. FISH analysis showed MDM2 gene amplification (Figure 1E). Hence, a final diagnosis of round cell sarcomas with EWSR1::PATZ1 fusion was rendered.

Patient 1. (A) Histophotomicrograph showing an undifferentiated tumor with cells arranged in sheets (H&E 100×). (B) High power showing tumor cells with round to spindled morphology with marked nuclear pleomorphism, coarse chromatin and few conspicuous nuclei (H&E 400×). (C) Immunohistochemistry showing focal nuclear immunoreactivity for S100 (DAB 200×). (D) Immunohistochemistry showing focal nuclear immunoreactivity for MYOD1 (DAB 400×). (E) FISH showing MDM2 amplification in the form of clusters of red signals (arrow) in few scattered tumor cells with CEP highlighted by green (1000×) (color figure online).
Given the aggressive nature of the disease and dearth of literature regarding effective treatment, he was initiated on adjuvant chemotherapy with ES-like protocol (dose-dense vincristine, adriamycin, cyclophosphamide (VAC) alternating with ifosfamide and etoposide (IE)). The patient completed 8 weeks of adjuvant chemotherapy and is doing well after 18 months of follow-up with no evidence of disease.
Patient 2
A 39-year-old man presented at another institute with complaints of fever, abdominal pain and an abdominal wall mass. Trucut biopsy from the lesion was reported as URCS. He subsequently underwent wide local excision, and histopathology revealed a malignant small round-to-spindled cell tumor (Figure 2A, B), morphologically suggestive of synovial sarcoma. The surgical margins were free but close.
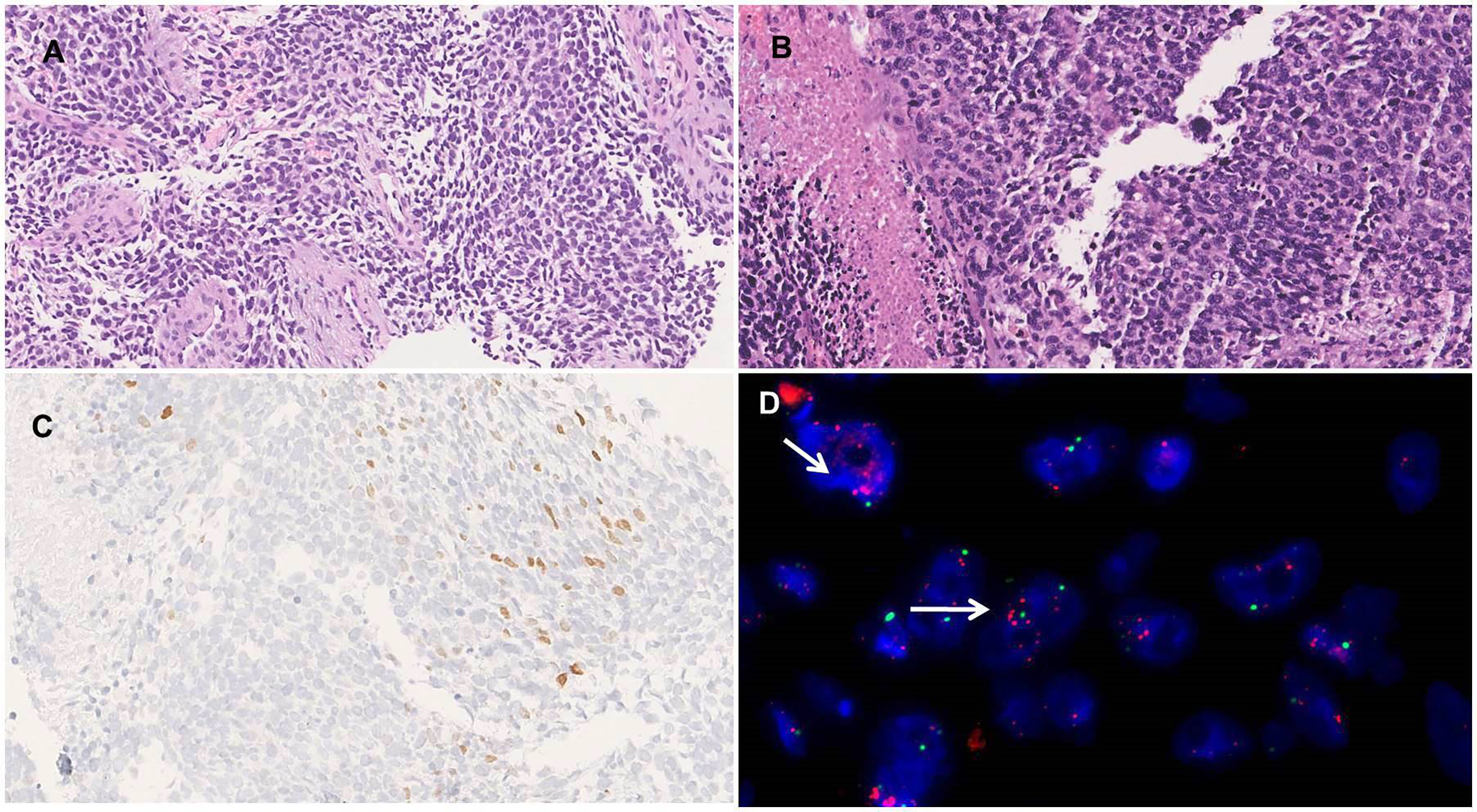
Patient 2. (A) Histophotomicrograph showing a malignant round to spindle cell tumor with intervening thick hyalinized vessels (H&E 200×). (B) High power showing cells with round to spindled morphology with moderate nuclear pleomorphism, vesicular chromatin, conspicuous nucleoli, moderate amount of cytoplasm, and areas of necrosis (H&E 400×). (C) Immunohistochemistry showing nuclear immunoreactivity for MYOD1 (DAB 400×). (D) FISH showing MDM2 amplification in the form of clusters of red signals (arrow) in tumor cells with CEP highlighted by green (1000×) (color figure online).
Review of biopsy blocks at two independent diagnostic laboratories showed varying diagnoses of RMS and de-differentiated liposarcoma with rhabdomyoblastic differentiation. These interpretations were based on immunopositivity for desmin, MDM2, MYOD1(focal) (Figure 2C), EMA (focal), and SMA (focal). The tumor was immunonegative for CD34, SOX10, CD99, NKX2.2, synaptophysin, pan-TRK, LCA, and CD30, with retained H3K27me3 immunoexpression. A FISH analysis showed MDM2 amplification (Figure 2D). A post-operative 18-fluorodeoxyglucose (FDG) PET-CT scan did not show metastatic or residual disease. He presented to our dedicated sarcoma clinic for further management. Due to the overlapping immunoprofile and discordant histopathology diagnosis, a sarcoma NGS panel was ordered, which revealed an EWSR1::PATZ1 fusion, between exon 8 of EWSR1 gene at the 5′ end and exon 1 of PATZ1 gene at the 3′ end. Hence, a final diagnosis of round cell sarcomas with EWSR1::PATZ1 fusion was rendered.
He was planned for adjuvant chemotherapy and radiotherapy. However, the patient defaulted and returned 6 months later, with a recurrence at the local site (Figure 3). FDG PET-CT scan showed an FDG avid lesion at the anterolateral aspect of the left lower abdominal wall, and no evidence of any loco-regional or distant metastasis. He was initiated on ES-based chemotherapy protocol (dose-dense VAC alternating with IE). The patient had local progression on chemotherapy and underwent surgery and adjuvant radiotherapy (60 gray in 30#). After 1 month of completion of radiation therapy, he developed widespread metastasis in lungs and brain (Figure 4A-C). The diagnosis was reconfirmed by NGS (FoundationOneCDx) which again revealed EWSR1::PATZ1 gene fusion, MDM2 amplification and CDKN2A deletion. He was started on immunotherapy (pembrolizumab) and trabectedin but progressed after two cycles. The poor prognosis was explained to the patient and his attendants, and palliative hippocampal avoidance whole-brain radiotherapy with simultaneous integrated boost was offered. He was subsequently lost to follow-up.
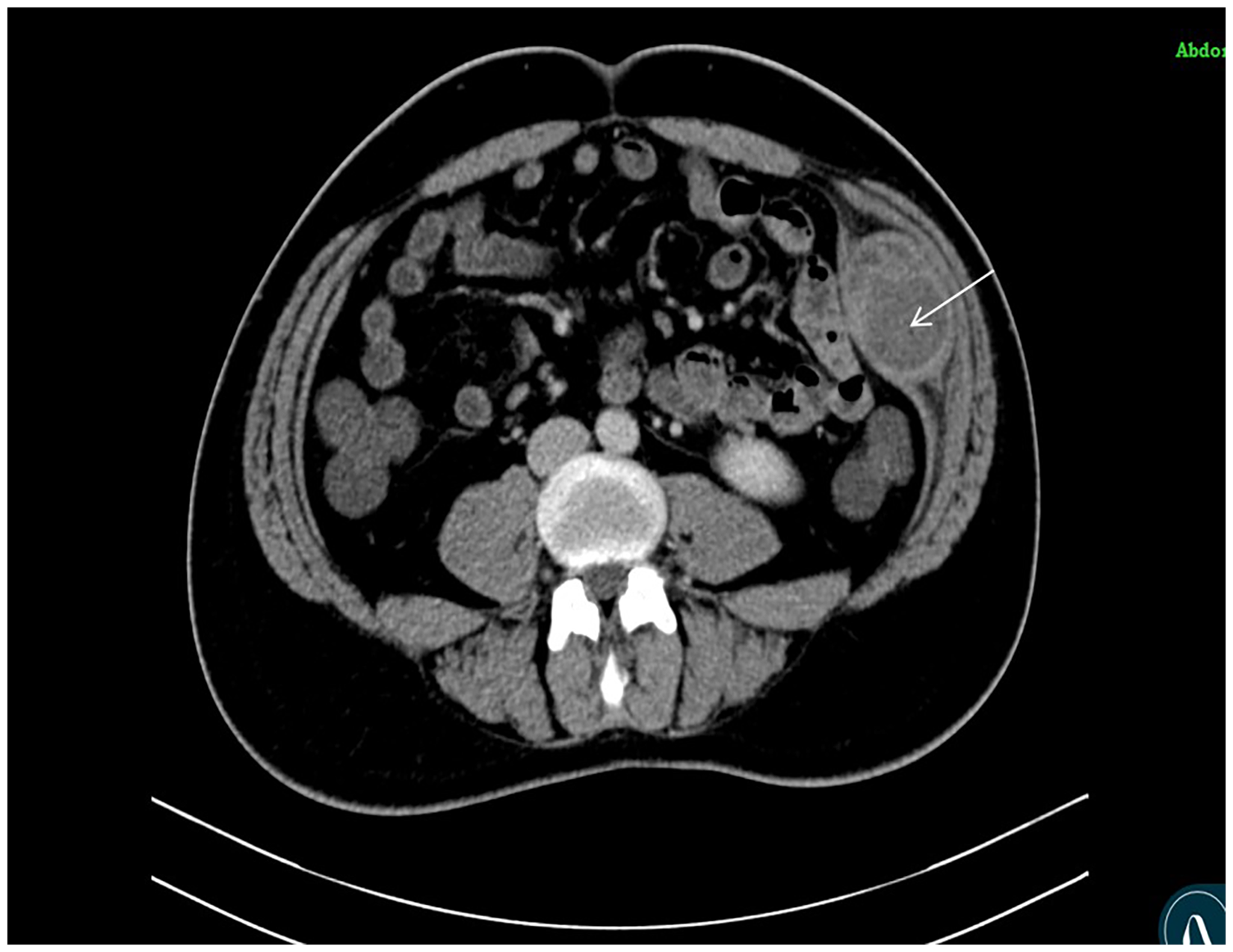
CT scan axial images showing well defined heterogeneously enhancing soft tissue density mass lesion (arrow) in intramuscular planes between internal oblique and transversus abdominis muscles.
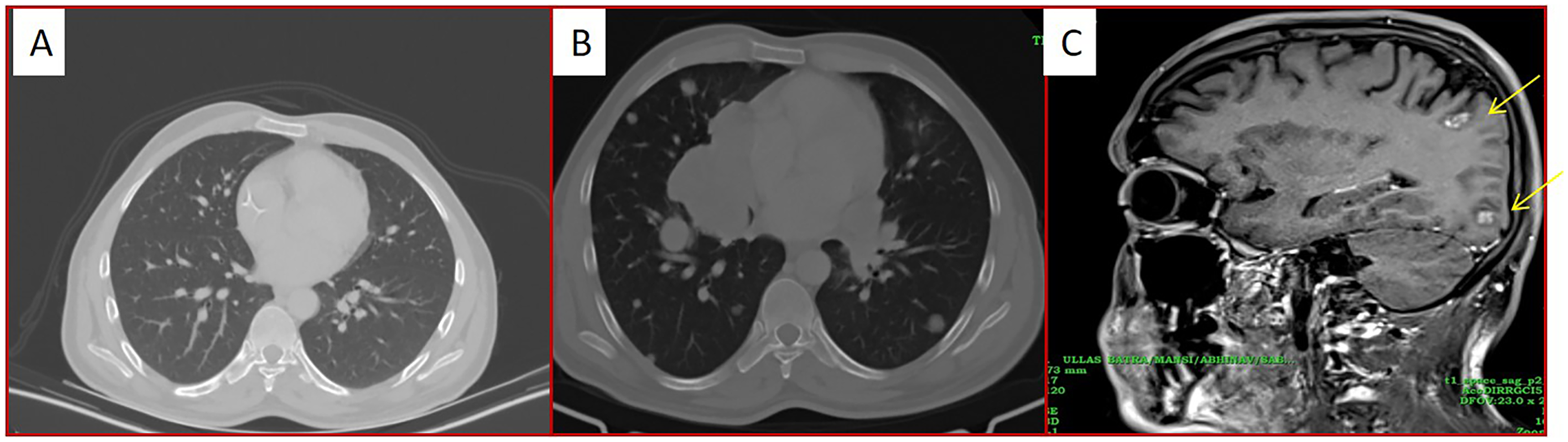
(A) Axial CT chest images done initially at the time of presentation show normal scan. (B) Axial CT chest after 3 months shows multiple metastatic lung nodules. (C) Sagittal post-contrast brain images show enhancing metastatic brain lesions (arrow).
Discussion
The fifth edition WHO classification of bone and soft tissue tumors includes a category of “undifferentiated round cell sarcomas (URCS) of bone and soft tissue.” 1 In addition to classical ES, it includes two distinct and separate entities: CIC rearranged sarcomas, and sarcomas with BCOR genetic alterations. The remaining tumors are grouped as “round cell sarcoma with EWSR1::non-ETS fusions.” The latter group includes 2 distinct gene fusions: EWSR1::NFATC2 and EWSR1::PATZ1. 1
Mastrangelo et al 7 described the first patient with EWSR1::PATZ1 fusion sarcoma more than two decades ago. Subsequent reports showed a wide age range from 1 to 68 years (median age of 42 years), slight male predominance, and a predilection for soft tissues of the chest wall and abdomen. Both of our patients were also located in the thoracoabdominal soft tissues. Grossly, these tumors often present as solid-cystic masses with variable necrosis and a tan yellow to grey centre. Microscopically, they consist of small round-to spindle cells in a fibrous stroma, with significant mitosis; however, can show a spectrum of histomorphology.2–4,7,8 Michal et al 4 have characterized the microscopic appearance into 3 categories: The first group is a low-grade tumor with round, ovoid, spindled, or epithelioid cells, absent necrosis, and hyalinized or myxo-hyaline stroma. The second group consists of round cells with minimal stromal matrix, and the third group is characterized by a high-grade spindle/round cell sarcoma with marked pleomorphism.
The immunoprofile of these tumors is polyphenotypic and often shows a distinctive co-expression of neural and myogenic markers. The tumors can show positivity for S100, GFAP, OLIG2 (neural markers), desmin, MYOD1, and PAX7 (myogenic markers). Immunoprofile varies from patient to patient. CD99 is seen occasionally; while EMA, BCOR, NKX2.2, NKX3.1, and STAT6 are usually negative. Due to the diverse clinical and pathological features, EWSR1::PATZ1 fusion sarcomas may mimic a multitude of sarcomas. These include MPNST, RMS, myoepithelial carcinoma, and other URCS. RMS is a close differential due to the histomorphology, and expression of myogenic markers as seen in our second patient was initially misdiagnosed as RMS.2–4,7,8
PATZ1 is a transcription factor regulating pluripotency and stemness. EWSR1::PATZ1 fusion leads to elimination of the N-terminal repressor domain leading to transcriptional dysregulation. Due to the proximity of PATZ1 to EWSR1 on chromosome 22, the EWSR1::PATZ1 fusion commonly results from sub-microscopic paracentric inversion and as a result, traditional BA-FISH may yield very low sensitivity. This has led to adoption of high-throughput NGS-based assays for diagnosis of EWSR1::PATZ1 fusion sarcoma, which helped in documentation of more patients in the prevailing literature. CDKN2A/CDKN2B loss has also been reported in up to 71% of these tumors and is believed to be associated with an aggressive course and poor prognosis.3,6,7,9 Concurrent MDM2 amplification has been reported in a subset of these tumors and is associated with poor response to therapy and an adverse prognosis.3,5 Patient 2 showed immunoexpression of MDM2 along with gene amplification espoused with immunoexpression of myogenic markers, which led to initial erroneous diagnosis of RMS and de-differentiated liposarcoma with rhabdomyoblastic differentiation.
EWSR1::PATZ1 sarcomas are generally considered to have an aggressive clinical course and modest response to chemotherapy.8,9 Recently, Dehner et al 8 analyzed 17 patients, of which 16 underwent complete surgical resection and 7 patient received neoadjuvant chemotherapy and radiation therapy. None of the evaluated tumor had any alterations of CDKN2A/B and/or TP53, or MDM2 amplification. The prognosis reported in their study was at variance in terms of existing literature. Twelve patients (75%) with clinical follow-up had no evidence of local or distant recurrence at a median follow-up duration of 13.5 months. The authors proposed that the prognosis of these rare tumors could be more favorable than previously reported as the clinical follow-up was not available for nearly half of the patients in previous reported series, thereby raising the distinct valid possibilities of reporting and/or referral bias. Moreover, obtaining clinical follow-up is often more difficult in patients in which tumors have behaved in a benign manner. Michal et al 4 have also suggested that at least some EWSR1::PATZ1 sarcomas may behave in an indolent fashion, although it is unclear whether any histopathological variables might be associated with better prognosis.
Management of EWSR1::PATZ1 fusion sarcomas is challenging given the heterogeneous presentation, aggressive clinical course, and absence of definite effective treatment options. Timely diagnosis, complete surgical resection with clear wide margins forms the mainstay of treatment, and response to chemotherapy is generally poor in the neoadjuvant setting.8,9 The effectiveness of adjuvant chemotherapy is still not clear owing to the handful of tumors reported in different case series. Data regarding chemotherapy regime is also limited, with a variety of chemotherapy regimens that have been tested in the neoadjuvant, adjuvant and metastatic testing. A majority of them have been treated on the lines of ES, with VAC or alternating VAC/IE. Other regimes include ifosfamide doxorubicin, carboplatin-paclitaxel, gemcitabine-docetaxel, pazopanib, and trabectedin.2–4,8–10 Many of the treated tumors showed a short-lived response, later followed by a recurrence or progression of disease.2–4,8–10
To conclude, we report two patients with rare EWSR1::PATZ1 fusion sarcomas as an attempt to add to the existing literature regarding their clinicopathological features. The immunoprofile is variable and overlaps with tumors showing rhabdomyoblastic or neural differentiation. Awareness of this entity, diligent histopathological evaluation, espoused with NGS-based molecular testing are essential for accurate diagnosis; the latter provides additional genetic information which can be utilized for prognostic and therapeutic information. Larger collaborative studies are required to validate whether any histopathological or molecular characteristics are attributed to prognostic and therapeutic significance.
Footnotes
Ethical Approval
Ethical approval for this study was waived.
Authors Contributions
S.P., S.R., D.B., Y.D., S.A.S., and A.B. did substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data.
S.P., S.R., and D.B. drafted the article and revised it critically for important intellectual content.
S.P., S.R., D.B., Y.D., S.A.S., and A.B. did final approval of the version to be published. All authors confirm they have meaningfully contributed to the research and read and approved the final manuscript.
Informed Consent
Written informed patient/next of kin consent was obtained for publication.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of Data and Material
Not applicable.
Presentation at any Meeting
Not applicable.
Trial Registration
Not applicable, because this article does not contain any clinical trials.