
Abstract
Adenosine deaminase deficiency causes severe combined immunodeficiency, affecting T and B cells and increasing the risk of infections, immune dysregulation, and malignancy. Undifferentiated embryonal sarcoma of the liver is a rare pediatric mesenchymal tumor. We report a 7-year-old boy with adenosine deaminase deficiency presenting with fever, elevated acute phase reactants, and an 85 × 60 mm2 heterogeneous cystic liver mass on ultrasonography. Magnetic resonance imaging revealed a second nodule in segments 5 and 6. An abscess was initially suspected, but biopsy confirmed undifferentiated embryonal sarcoma of the liver. After chemotherapy, the patient underwent a left lobectomy and wedge resections. Histology showed complete necrosis of the sarcoma; however, a second tumor—fibrolamellar hepatocellular carcinoma—was identified. While immunodeficiencies are associated with lymphoproliferative disorders and malignancies, the link between adenosine deaminase deficiency and hepatic tumors is unclear. This patient's presentation highlights the potential for multiple primary hepatic malignancies in immunodeficient patients and suggests a need for further investigation.
Keywords
Introduction
Adenosine deaminase is a critical enzyme in the purine degradation and salvage pathway. Adenosine deaminase defects cause adenosine, deoxyadenosine, and total deoxyadenosine nucleotides to build up. This accumulation impairs cell signaling processes, including energy production and DNA repair. Patients with adenosine deaminase deficiency frequently experience severe combined immunodeficiency, a condition that affects both T and B lymphocytes, as the latter are particularly susceptible to the effects of adenosine deaminase deficiency. An elevated risk of infection, immune dysregulation, malignancy, and infant mortality is associated with severe combined immunodeficiency. 1 Although survival has improved in the last decade due to improved therapeutic procedures such as enzyme replacement therapy and stem cell transplantation, this has led to an overall increase in the risk of developing malignancies (estimated at 4%-25%). Non-Hodgkin lymphoma is the most common malignancy seen in patients with immunodeficiency, accounting for 60% of malignancies in this population. 2 In addition to lymphoma, patients with adenosine deaminase deficiency-associated severe combined immunodeficiency are at a higher risk of developing multicentric dermatofibrosarcoma protuberans.3,4 It is also thought that the use of polyethylene glycol-conjugated adenosine deaminase replacement therapy beyond 10 years may also result in an increased risk of lymphoma. For instance, Kaufman et al 5 reported a patient who developed a cerebral lymphoma after receiving polyethylene glycol-conjugated adenosine deaminase therapy for 10 years. Husain et al 6 also described the development of Burkitt's lymphoma in a patient who received polyethylene glycol-conjugated adenosine deaminase for 13 years. However, the relationship between adenosine deaminase deficiency-associated severe combined immunodeficiency and nonhematopoietic malignancies is not clear. To our knowledge, only 1 instance of hepatocellular carcinoma has been reported in an 8-month-old adenosine deaminase deficiency-associated severe combined immunodeficiency patient, occurring after successful hematopoietic stem cell transplantation. 7
Primary malignant liver tumors are rare in children and the most common type is hepatoblastoma, followed by hepatocellular carcinoma. Embryonal sarcoma is the third most common malignant liver tumor, making up 6% to 13% of all malignant pediatric liver tumors. Although most tumors are sporadic, malignant transformation arising from mesenchymal hamartoma has been described. 8 Here, we present a patient with adenosine deaminase deficiency-associated severe combined immunodeficiency diagnosed with synchronous undifferentiated embryonal sarcoma of the liver and fibrolamellar hepatocellular carcinoma.
Case Report
A 7-year-old boy, who was diagnosed with adenosine deaminase enzyme deficiency in our center when he was 18 months old and underwent hematopoietic stem cell transplantation from an unrelated donor when he was 30 months old, was referred to our center from another hospital because of a fever, elevated acute phase reactants, and the detection of an 85 × 60 mm2 heterogeneous mass with cystic content in the left liver lobe on abdominal ultrasonography. The parents were first-degree consanguineous, and the mother had a history of 2 prior abortions and 2 spontaneous pneumothoraxes, 1 of which developed during the pregnancy of our patient. There was no family history of immunodeficiency or early child loss. Upon examination, the patient's body weight was 28 kg (90th percentile), height was 120 cm (35th percentile), head circumference was 53 cm (80th percentile), and no abnormal findings were detected in the systemic examination. In the left lobe of the liver, a heterogeneous, solid mass with cystic necrotic areas measuring 87 × 80 × 50 mm3 was observed during the ultrasonography procedure conducted at our center. Several other echogenic solid lesions measuring 11 × 13 mm2 were observed in the right lobe of the liver, with the largest one located at the level of segment 6. There was no abnormality in the patient's complete blood count and biochemistry parameters, alpha-fetoprotein was 1.13 ng/mL (N), C-reactive protein was 140 mg/L, and sedimentation was 102 mm/h.
Thereupon, an abdominal magnetic resonance imaging was performed for the differential diagnoses of abscess and malignancy. In axial postcontrast hepatobiliary phase magnetic resonance images, a heterogeneous enhanced mass, measured 82 × 74 × 52 mm3 in the left lobe of the liver and a hypointense nodular lesion sized 19 × 16 mm2 in segment 6 of the right lobe were observed (Figures 1 and 2). Considering the patient's primary immunodeficiency, granulomatous lesions or abscess formation were considered more likely. Hepatoblastoma was also considered in the differential diagnosis. Portal lymph nodes were observed, and a tissue diagnosis was advised.
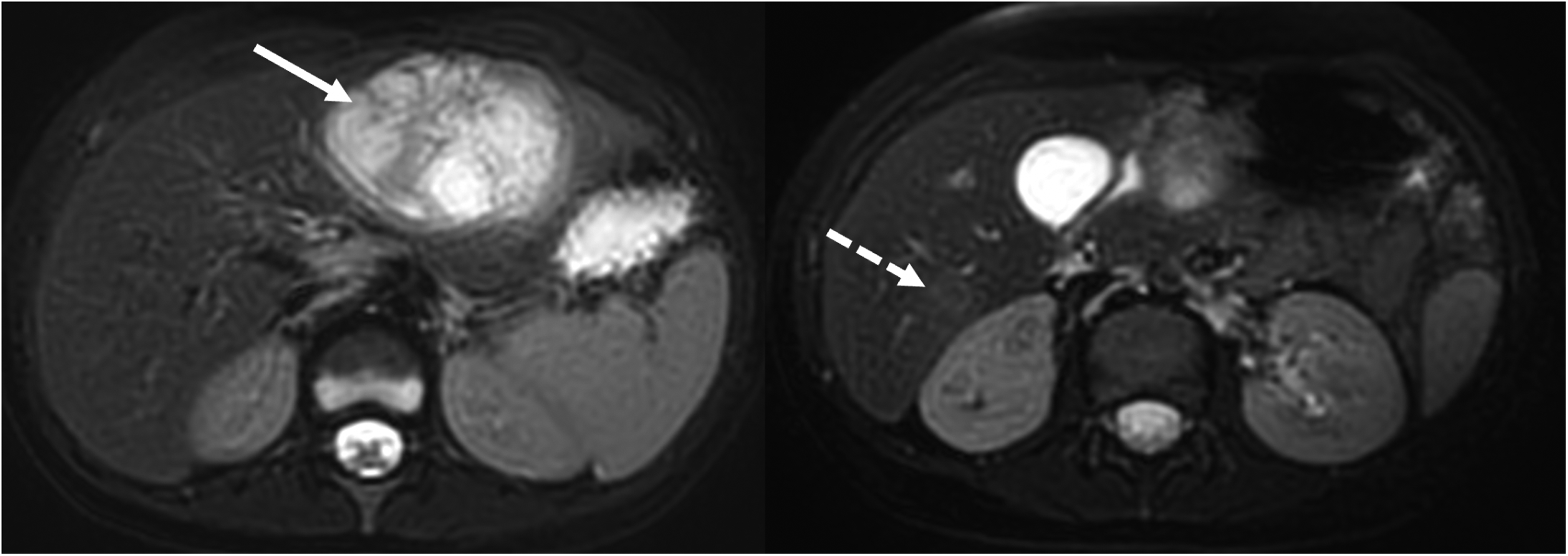
Axial T2-weighted magnetic resonance images show a heterogeneous hyperintense mass in the left lobe (arrow). Note the slightly hyperintense lesion in the right lobe (dashed arrow).
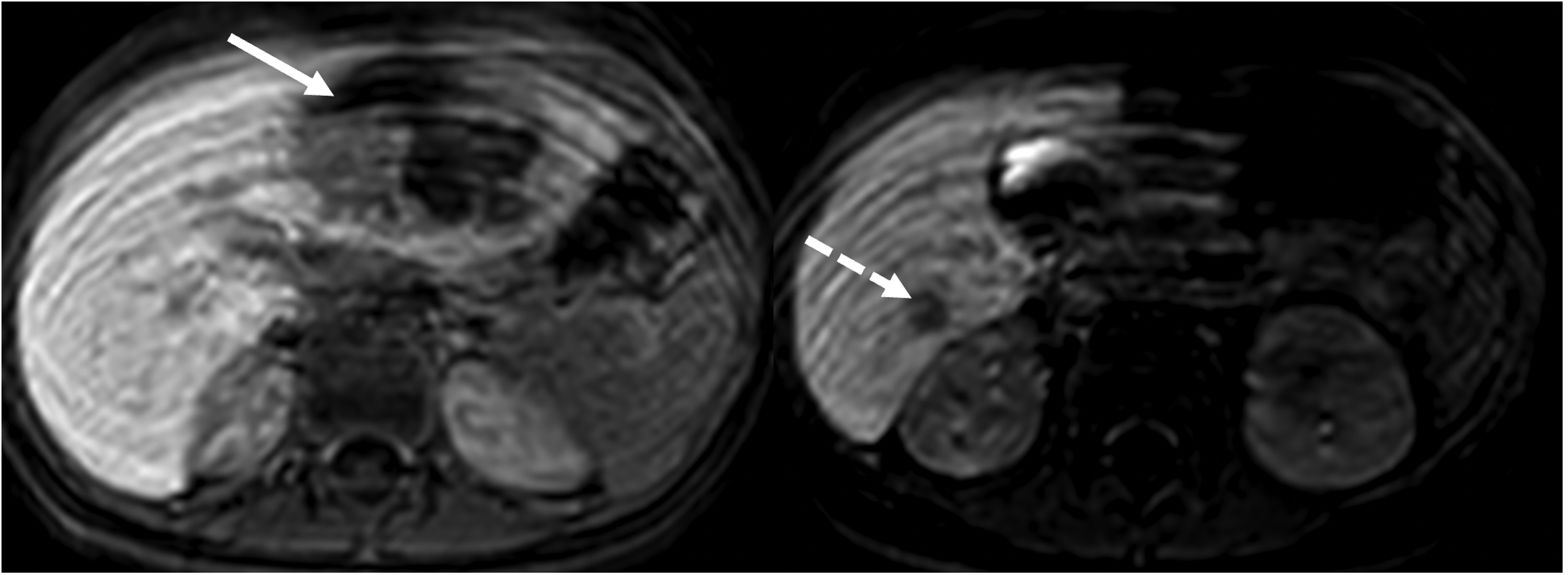
Axial postcontrast hepatobiliary phase magnetic resonance images demonstrate a heterogeneous enhanced mass in the left lobe (arrow) and a hypointense nodular lesion in the right lobe (dashed arrow).
The patient underwent image-guided core needle biopsy by the interventional radiology department, sampling only the larger mass seen in the left hepatic lobe. Part of the tissue sample was sent for aerobic culture, fungal culture, and tuberculosis panel. The histopathologic examination of the core biopsy revealed a neoplasm composed of spindle/stellate cells embedded in a myxoid stroma. The neoplastic cells had hyperchromatic pleomorphic nuclei, granular eosinophilic cytoplasm, and characteristic periodic acid-Schiff (PAS)-positive intracytoplasmic globules. Bizarre giant cells and areas of necrosis were also present. Immunohistochemically, the neoplastic cells were only positive with desmin. Pan-keratin (AE1/3), HepPar-1, Myo-D1, myogenin, and glypican-3 were negative (Figure 3). With these findings, the diagnosis of undifferentiated embryonal sarcoma of the liver was made. The patient was transferred to the oncology department. Positron emission tomography-computed tomography (PET-CT) performed by the nuclear medicine department for staging before treatment was reported as a malignant mass with heterogeneous fluorodeoxyglucose (FDG) uptake in the left lobe of the liver and left anterior diaphragmatic and abdominal lymph nodes with increased FDG uptake. The lesion in segment 6 was not mentioned in the PET-CT report. The patient received 3 cycles of ifosfamide–adriamycin chemotherapy and the mass in the left lobe of the liver was observed to have marginally shrunk in the CT scan. However, the hypodense area in segment 6 of the right lobe remained the same in size. Two months later, the patient underwent left hepatectomy and wedge resection of segments 5 and 6. Macroscopically, the cut section of the left liver lobe revealed a solid/necrotic mass measuring 7.5 × 5.2 × 4.5 cm (Figure 4). The wedge resection from segment 5 showed a white solid mass measuring 2.3 × 1.4 × 1.4 cm, while no macroscopic mass was identified in the cut surface of the segment 6 wedge resection. In the microscopic examination, the mass initially diagnosed as embryonal sarcoma in the left lobe of the liver was diffusely necrotic and only a few viable neoplastic cells were identified. In segments 5 and 6, however, a completely different tumor composed of large polygonal cells with vesicular chromatin, prominent nucleoli, and abundant eosinophilic cytoplasm was identified (Figure 5). These neoplastic cells were arranged in trabeculae/cords and nests in a dense fibrous stroma. The morphologic diagnosis of fibrolamellar hepatocellular carcinoma was confirmed with immunohistochemical examination. The neoplastic cells were positive with HepPar-1 and keratin 7 (KRT7), which were both negative in the embryonal sarcoma in the left lobe. CD68 showed patchy cytoplasmic staining. With these morphological and immunohistochemical findings, these tumor nodules were diagnosed as fibrolamellar variant of hepatocellular carcinoma.

(A) Hyperchromatic, pleomorphic spindled neoplastic cells in a myxoid stroma with scattered giant bizarre cells (hematoxylin–eosin stain, 200×). (B) Characteristic PAS-positive eosinophilic globules can be seen in the cytoplasm of neoplastic cells (PAS stain, 200×). (C) Neoplastic cells showing variable positivity with desmin (200×) immunohistochemistry. (D) No staining was seen with HepPar-1 immunohistochemistry (200×).

(A) Macroscopic examination of the left liver lobectomy showed a necrotic mass measuring 7.5 × 5.2 × 4.5 cm3. (B and C) Microscopic examination revealed near-complete necrosis in the embryonal sarcoma (hematoxylin–eosin stain, 3×, 40×).

(A, B, C) Wedge resection of segment 5 showed a different neoplasm (fibrolamellar hepatocellular carcinoma) made up of cells with prominent nucleoli, ample eosinophilic cytoplasm separated by fibrous bands (hematoxylin–eosin stain, 100× and 200×). (D) The neoplastic cells were diffusely positive with HepPar-1 immunohistochemistry (100×). (E) Keratin 7 was also diffusely positive (200×). (F) CD68 showed patchy cytoplasmic staining (40×).
Discussion
Undifferentiated embryonal liver sarcomas are characteristically seen in children aged 6 to 9 years and make up 9% to 15% of pediatric liver malignancies. 9 They are thought to originate from a developmental arrest of mesenchymal cells during embryonal development and the most prominent alteration is a structural rearrangement in chromosome 19q13, which is also typical for benign mesenchymal hamartoma. It has been hypothesized that a second event (such as TP53 mutation) may lead to malignant transformation. 10 Imaging in undifferentiated embryonal sarcoma of the liver typically reveals a large hepatic mass composed of both solid and cystic components, occasionally involving the portal system. Although there are no predisposing factors for the development of embryonal sarcoma, there are reports of germline p53 mutations in several patients. 9
Fibrolamellar hepatocellular carcinoma accounts for 15% to 25% of pediatric hepatocellular carcinomas. Unlike classic hepatocellular carcinoma, fibrolamellar hepatocellular carcinoma is seen in older children and patients typically do not have underlying liver disease. 8 This tumor is characterized by the DNAJB1::PRKACA fusion, which is specific in the setting of a liver neoplasm. 11 However, this fusion has also been identified in oncocytic and biliary neoplasms. A group of these tumors do not show this fusion, but have inactivating mutations of BAP1 (BRCA1-associated protein-1) gene instead. 12
The relationship between adenosine deaminase deficiency, embryonal sarcoma, and fibrolamellar hepatocellular carcinoma is unclear. Different mesenchymal tumors, such as giant cell fibroblastoma, 13 dermatofibrosarcoma protuberans,3,4 and mesenchymal hamartoma, 14 have been reported in patients with adenosine deaminase deficiency. In addition to mesenchymal tumors, Ucku et al reported the first case report of an 8-month-old adenosine deaminase deficiency-associated severe combined immunodeficiency patient who developed progressive liver dysfunction and hepatocellular carcinoma after successful hematopoietic stem cell transplantation. They emphasized a complex etiology of the liver dysfunction. 7 However, no reports of synchronous embryonal sarcoma and hepatocellular carcinoma have been described in literature. Li et al reported a concomitant hepatocellular carcinoma and embryonal rhabdomyosarcoma in a 40-year-old man with a history of hepatitis B infection. 15 To the best of our knowledge, synchronous embryonal sarcoma and fibrolamellar hepatocellular carcinoma have not previously been reported in a patient with adenosine deaminase deficiency-associated severe combined immunodeficiency.
The pathogenetic mechanisms underlying these 2 primary tumors and the role of immunodeficiency are not clear. The molecular hallmark of fibrolamellar hepatocellular carcinoma is the DNAJB1::PRKACA fusion, which occurs sporadically and is not linked to known hereditary tumor syndromes. No prior reports have described an association between adenosine deaminase deficiency and fibrolamellar hepatocellular carcinoma or embryonal sarcoma. Therefore, the coexistence in this patient may represent a coincidental finding rather than a manifestation of a shared pathogenic mechanism. Nevertheless, the chronic immune dysregulation and altered cellular metabolism characteristic of adenosine deaminase deficiency could theoretically contribute to tumorigenesis and warrant further investigation in future studies.
Thus, further research is needed to fully understand whether there is a link between adenosine deaminase deficiency and embryonal sarcoma or hepatocellular carcinoma and the underlying mechanisms that may contribute to tumor development in this population.
Footnotes
ORCID iDs
Ethical Considerations
Formal ethics committee approval was not required as this a single anonymized case report.
Consent to Participate
Written informed consent was obtained from the patient's parents.
Author Contributions
AN: conceptualization, manuscript writing, and pathological assessment. AK: manuscript writing and clinical assessment. DIG: manuscript writing and clinical assessment. ED: manuscript writing and clinical assessment. HSS: clinical assessment and manuscript editing. HNO: radiological assessment, manuscript writing, and manuscript editing. EY: manuscript writing and clinical assessment. SE: clinical assessment and supervision. DC: clinical assessment, manuscript editing, and supervision. DO: pathological assessment, manuscript editing, and supervision.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.