
Abstract
Primary pulmonary myxoid sarcoma (PPMS) is an extremely rare malignant pulmonary neoplasm. Its pathogenesis is closely linked to the chromosomal translocation t(2;22)(q33;q12), which drives the formation of the EWSR1::CREB1 gene fusion. This pathognomonic genetic alteration serves as one of the key clues for the pathological diagnosis of PPMS. In this paper, we report a patient with PPMS, a 64-year-old female patient, in whom the EWSR1::CREB1 gene fusion was definitively confirmed via fluorescence in situ hybridization. The tumor had been present for 7 years prior to resection without causing symptoms, suggesting an indolent growth pattern. The patient received no adjuvant therapy and remained without evidence of disease at an interval of 46 months following surgery. The main value of this report is an additional patient with a detailed documentation of indolent behavior, both pre-operatively and post-operatively.
Introduction
Primary pulmonary myxoid sarcoma (PPMS) is an exceedingly rare mesenchymal neoplasm first described by Thway et al (2011). 1 To date, the exact etiology of this entity remains elusive. Some expert soft tissue pathologists regard PPMS as representing the same pathological entity as AFH. 2 The myxoid variant of AFH was characterized subsequent to the initial description of PPMS, as documented by Schaefer and Fletcher, 3 which accounts for why this association was not recognized at the outset.
Approximately 80% of tumors are defined by a distinct genetic aberration, specifically the EWSR1::CREB1 gene fusion. In the 2021 WHO Classification of Thoracic Neoplasms, this entity was officially designated as PPMS with EWSR1::CREB1 fusion.4,5 Originating predominantly from the mesenchymal tissue of the airways, PPMS typically manifests an endobronchial, lobulated growth pattern. Affected patients may present with symptoms such as cough or hemoptysis, accompanied by signs of bronchial obstruction. Characterized by intermediate biological behavior, PPMS has a median age of onset of approximately 45 years. Radiological assessments, including computed tomography (CT) scans and chest radiographs, usually identify peribronchial pulmonary masses. 6 In terms of gross morphology, PPMS tumors are generally less than 4 cm in diameter, featuring well-circumscribed margins and a nodular configuration. On cross-section, the tumors exhibit a myxoid, gelatinous texture and are predominantly yellowish or whitish in color. Definitive diagnosis relies on the detection of EWSR1::CREB1 fusion, which can be achieved via techniques such as fluorescence in situ hybridization (FISH) or next-generation sequencing. Currently, PPMS is categorized as a low-grade malignant neoplasm.7,8 The clinical course is typically indolent in most patients, with the majority showing no evidence of recurrence or metastasis following surgical resection.
Clinical History
A 64-year-old female patient was incidentally detected with a pulmonary mass during a routine physical examination at a local hospital 7 years prior before surgery, for which no treatment was administered at that time. At the time of presentation, she had a follow-up visit at the outpatient department and underwent an enhanced CT scan. Imaging findings showed increased pulmonary markings; a well-circumscribed, nodular soft tissue density shadow, measuring approximately 1.8 cm × 1.5 cm, was identified in the right upper lobe, with heterogeneous enhancement following contrast medium administration. Scattered linear opacities were also observed bilaterally. The trachea and main bronchi remained patent, and no prominent lymphadenopathy was noted in the mediastinal or hilar regions. Furthermore, there were no signs of pleural effusion in either thoracic cavity (Figure 1). Throughout the disease course, the patient had no other specific symptoms, and her past medical history was unremarkable.
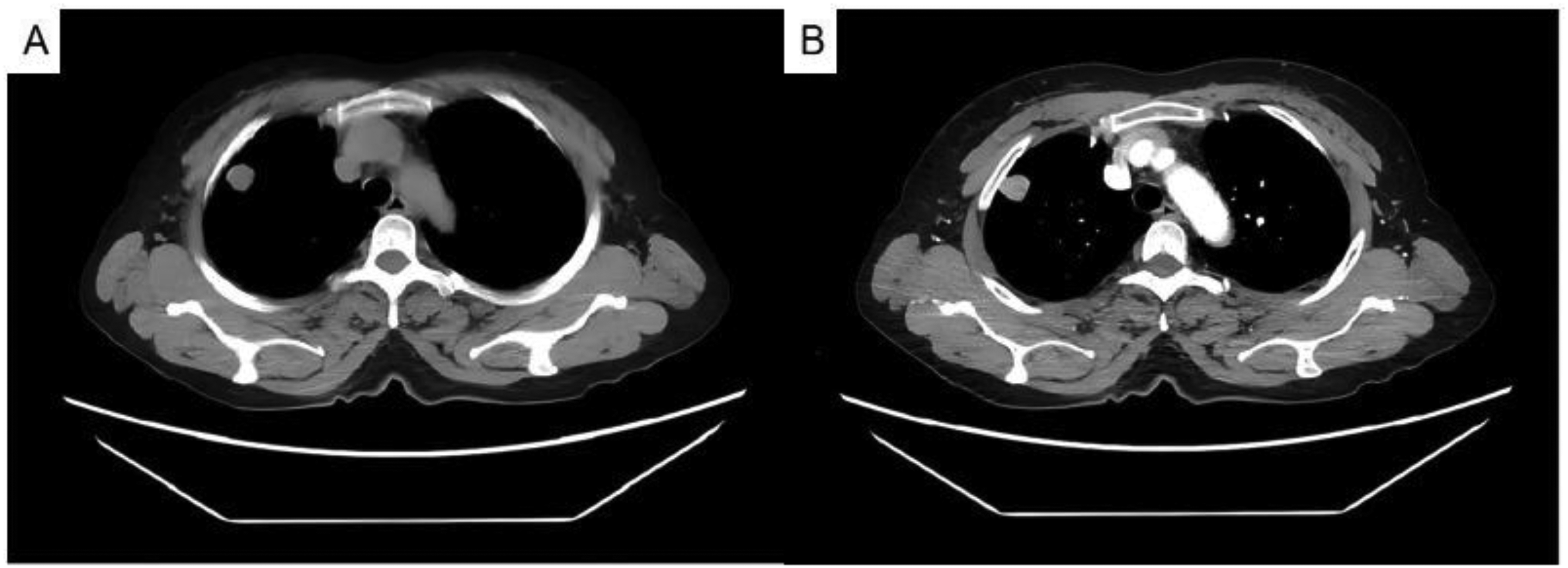
Ct findings demonstrated a well-circumscribed, nodular soft tissue density shadow in the right upper lobe, measuring approximately 1.8 cm × 1.5 cm. The lesion showed heterogeneous enhancement after contrast medium administration. (A) plain scan; (B) arterial phase.
Although the lesion remained radiologically stable and asymptomatic during the 7-year observation period, the patient eventually opted for surgical resection to obtain a definitive histopathological diagnosis and exclude malignancy. A week later, the patient was scheduled for thoracoscopic resection of the right upper lobe lesion under general anesthesia. Intraoperatively, a firm, well-circumscribed lesion with dimensions of approximately 2.0 cm × 1.5 cm was localized in the right upper lobe. The overlying pleura of the lesion presented with prominent protrusion, while no adhesions to the thoracic wall were detected. Moreover, no notable enlargement of paratracheal, hilar, or mediastinal lymph nodes was identified, and no pleural effusion was evidenced. Intraoperative frozen section analysis preliminarily suggested a probable spindle cell tumor in the right upper lobe, though a conclusive diagnosis remained contingent on the outcomes of routine pathological assessment and immunostaining assays. A wedge-shaped segment of lung tissue with dimensions of 7 cm × 2 cm × 1.5 cm was observed. A solitary mass measuring 2 cm × 1.5 cm × 1.5 cm was detected within the specimen, situated 2 cm from the surgical resection margin and close to the visceral pleura but not involving it. The sectional surface of the mass displayed a grayish-white appearance and a soft, gelatinous consistency. Under the microscope, the tumor cells are spindle-shaped with scarce cytoplasm and mild atypia, scattered in a myxoid matrix; the tumor cells grew in nodules, surrounded by fibrous pseudocapsules. Subsequent definitive pathological analysis verified the lesion as a spindle cell tumor with prominent myxoid features (Figure 2).

Representative hematoxylin and eosin (H&E)-stained sections of the pulmonary tumor demonstrate the following distinct histological characteristics: (A) low-magnification view (×40) revealing a well-defined fibrous pseudocapsule encircling the tumor mass; (B) medium-magnification view (×100) displaying abundant mucoid stroma within the tumor; (C) high-magnification view (×400) depicting tumor cells arranged primarily in cord-like configurations, with heterogeneous morphologies including spindle, stellate, and polygonal forms; (D) high-magnification view (×400) showing the presence of lymphocytes and plasma cells at the tumor margin.
Immunohistochemical staining assays were conducted via the EnVision technique, and the expression patterns of a panel of tumor markers were evaluated. The results demonstrated the following immunoreactivity profiles: Positive immunoreactivity: EMA (partial positivity), CD68 (partial positivity, expressed in neoplastic cells), SMA (focal positivity, in tumor cells and intra-tumoral vascular), desmin (focal positivity), calponin (focal positivity), and Ki-67(MKI67) (index: 1%); negative immunoreactivity: pankeratin, KRT7, KRT18, TTF-1 (NKX2-1), CD34, ALK (clone D5F3, Ventana), S100, GFAP, CD21, CD38, CD123, SYP (synaptophysin), CHGA (chromogranin A), and P16.
Based on histomorphological and immunohistochemical features, this lesion could not reliably differentiated from PPMS, AFH, and extraskeletal myxoid chondrosarcoma. Further molecular detection assays targeting the EWSR1::CREB1 fusion gene are strongly recommended to establish a definitive diagnosis. FISH assays targeting the EWSR1::CREB1 fusion gene were performed on the patient's specimen, utilizing the LBP EWSR1::CREB1 fusion gene rearrangement probe (Guangzhou LBP Medicine Science & Technology Co., Ltd, Guangzhou, China). Molecular testing results definitively verified the existence of the recurrent EWSR1::CREB1 gene fusion event (Figure 3). Integrating these molecular diagnostic findings with histopathological morphology and clinical manifestations, the definitive diagnosis in this patient was established as PPMS.

Representative immunohistochemical staining and FISH results of the pulmonary tumor, illustrating key pathological features of PPMS harboring the EWSR1::CREB1 fusion gene. (A) EMA (focal positive); (B) desmin (focal positive); (C) ALK(clone D5F3, Ventana); (D) EWSR1::CREB1 fusion gene detection, FISH. PPMS, primary pulmonary myxoid sarcoma; FISH, fluorescence in situ hybridization.
Follow-up
The patient did not receive adjuvant therapy and remained disease-free at 46 months post-surgery. No adjuvant radiation or chemotherapy was administered following surgical resection.
Discussion
PPMS is an exceedingly rare malignant mesenchymal neoplasm that arises from the airway structures. A defining pathological feature of this entity is the presence of the EWSR1::CREB1 gene fusion, which is detected in the vast majority of documented tumors. 9 EWSR1 (Ewing sarcoma breakpoint region 1), located on chromosome 22q12, was first identified in Ewing sarcoma. EWSR1 belongs to the FET family of RNA-binding proteins, which also includes FUS and TAF15. FET::CREB fusion-positive neoplasms constitute a spectrum of mesenchymal tumors characterized by fusions between FET family members and CREB family transcription factors (CREB1, ATF1, and CREM), including angiomatoid fibrous histiocytoma, clear cell sarcoma, and PPMS. 10 Accumulating evidence indicates that the EWSR1::CREB1 rearrangement serves as a critical oncogenic driver contributing to the initiation and progression of malignant neoplasms harboring this aberration. 11
Recently, the spectrum of FET::CREB fusion-positive neoplasms has expanded to include FET::CREM fusion mesenchymal neoplasms, which have been identified at both extra-abdominal and intra-abdominal sites. 12 These tumors demonstrate potentially aggressive clinical behavior and share overlapping morphological and immunophenotypic features with other FET::CREB fusion-positive entities, highlighting the expanding family of mesenchymal neoplasms driven by FET::CREB family fusions. This emerging entity underscores the importance of comprehensive molecular profiling in the diagnostic workup of myxoid mesenchymal neoplasms.
Histopathologically, PPMS characteristically displays an endobronchial growth pattern with protrusion into the bronchial lumen, and it may further infiltrate the surrounding lung parenchyma in advanced stages. With respect to epidemiological characteristics, PPMS affects both sexes with nearly equal frequency, albeit with a slight predilection for middle-aged female patients. The age range at diagnosis is broad, spanning from 21 to 80 years old. Clinically, PPMS manifests with non-specific clinical presentations, among which cough is the most prevalent symptom. Other associated manifestations may include expectoration, hemoptysis, or systemic manifestations such as unintentional weight loss. 13 Radiological findings are similarly non-specific, chest CT scans predominantly demonstrate intrapulmonary space-occupying lesions, and bronchial obstruction is a common complication due to the endobronchial growth of the tumor. From a biological perspective, PPMS generally exhibits an indolent clinical behavior, marked by slow tumor proliferation and limited metastatic dissemination, as supported by the 7-year asymptomatic preoperative course observed in this patient.
Pathological examination serves as the gold standard for the definitive diagnosis of PPMS, with the disease exhibiting the following distinct histological and molecular characteristics:① Cytological characteristics: Cytological features are non-specific. Tumor cells usually exhibit a spindle-to-epithelioid morphology, accompanied by moderate volumes of myxoid cytoplasm.② Histological characteristics: Tumors typically manifest as lobulated or nodular masses, composed of hypocellular and myxoid regions partitioned by dense fibrous vascular bands. The tumor is often closely associated with the bronchial wall, and neoplastic cells are arranged in reticular, nested, and cord-like architectures, embedded within an abundant loose myxoid stroma. Cellular atypia is mild to minimal, mitotic activity is low (typically fewer than 5 mitotic figures per 2 mm2), and necrosis is predominantly focal when present.③ Immunohistochemical profile: Tumor cells show occasional focal positivity for EMA, 1 and are negative for keratin, CD34, and neuroendocrine markers, with variable focal positivity for desmin and SMA.④ Molecular genetic characteristics: Molecular testing reveals that approximately 80% of PPMS tumors harbor EWSR1::CREB1 gene rearrangement, which represents a key diagnostic hallmark of the disease.
For the differential diagnosis of PPMS, it is critical to distinguish this entity from other pulmonary spindle cell neoplasms that exhibit myxoid stromal features. Such lesions include: AFH, pulmonary hamartoma, chondroma, myoepithelial tumor, inflammatory myofibroblastic tumor, epithelioid hemangioendothelioma, pulmonary myxoma, invasive vascular myxoma, radiation-induced sarcoma, primary pulmonary myxosarcoma, and metastatic sarcomas (eg, myxoid liposarcoma and extraskeletal myxoid chondrosarcoma).14–18 Extraskeletal myxoid chondrosarcoma is a rare, malignant mesenchymal neoplasm of uncertain histogenetic origin. Although it most frequently arises in the deep soft tissues of the lower extremities, extramusculoskeletal involvement—including pleural and other thoracic sites—has been increasingly documented. Clinically, extraskeletal myxoid chondrosarcoma typically manifests as a slow-growing, well-circumscribed mass; gross examination reveals a gelatinous, translucent cut surface. Histopathologically, the tumor exhibits a characteristic lobular architecture, with uniform oval to short spindle-shaped cells arranged in cords, clusters, or small nests embedded within an abundant, hypocellular myxoid stroma; notably, true hyaline cartilage differentiation is absent. At the molecular level, extraskeletal myxoid chondrosarcoma is defined by recurrent rearrangements involving the NR4A3 gene, most commonly resulting in fusion transcripts such as EWSR1::NR4A3, TAF15::NR4A3, TCF12::NR4A3, or TFG::NR4A3. This distinct genetic profile reliably differentiates extraskeletal myxoid chondrosarcoma from PPMS, which harbors EWSR1::CREB1 fusions. PPMS and AFH exhibit substantial overlap in histological morphology, clinical manifestations, immunophenotypic signatures, and molecular alterations. Notably, the recent identification of the EWSR1::ATF1 fusion in a subset of PPMS tumors has raised the provocative hypothesis that PPMS and AFH may represent morphological variants of a single tumor entity, as proposed by Kerper et al. 2 While the similar genetics, morphology, and IHC profiles could support that PPMS represents a site-specific form of AFH, more study is needed to better understand the relationship between these tumor types.
PPMS demonstrates intermediate biological behavior, which is typified by indolent tumor growth and occasional distant metastatic potential. Owing to the extreme rarity of this entity and the lack of pathognomonic clinical manifestations, no validated prognostic factors have been identified as of the last follow-up. Surgical resection is generally regarded as the first-line therapeutic approach for resectable lesions and is associated with relatively favorable clinical outcomes. However, long-term regular follow-up is imperative to surveil for disease recurrence or distant metastasis.
Footnotes
Acknowledgments
We acknowledge the technical support from the Affiliated Hospital of Nantong University and it's pathology.
Ethical Approval
The study was approved by the Ethics Committee of the Affiliated Hospital of Nantong University 2020-K007.
Author contributions
Conceptualization was done by YL and QZ; methodology was done by YL and QZ; software was done by YS and QZ; validation by YL and QZ; formal analysis by all authors; investigation by QZ; resources by YL; data curation by QZ; writing—original draft by all authors; writing—review and editing by all authors; visualization by QZ; supervision by QZ; project administration by QZ; funding acquisition by QZ.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by grants from Basic Science Research Project in Nantong City, Jiangsu, China (JC2021029). Project Title: Investigation of the Molecular Mechanisms by Which the DDX46/FTO/BCL-2/Beclin1 Signaling Axis Regulates Autophagy and Proliferation in Lung Adenocarcinoma.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.