
Abstract
Cancer-associated pain is common, debilitating, and increasingly recognized not merely as a symptom but as an active participant in disease progression. Its etiology reflects a dynamic convergence of tumor-derived secretory factors, therapy-induced biochemical alterations, and a persistently inflamed tumor microenvironment (TME). Neuropilin-1 (NRP1), a transmembrane co-receptor with broad ligand specificity, has emerged as a key integrator of these diverse pro-nociceptive signals. This review examines how NRP1 coordinates pain pathways triggered by tumor-secreted mediators—including nerve growth factor (NGF), vascular endothelial growth factor A (VEGFA), and hepatocyte growth factor (HGF). We narratively reviewed its role in assembling signaling complexes, particularly with NGF and TrkA, that amplify nociceptive transduction. The therapeutic rationale for targeting NRP1 with biologic and small-molecule inhibitors is analyzed, with emphasis on combining these agents with conventional opioids. Such combination strategies aim to achieve multi-mechanistic analgesia, reduce opioid-related side effects, and interrupt the reciprocal reinforcement between pain signaling and the TME. Finally, we narratively reviewed translational progress, identify persistent challenges, and outline future directions for NRP1-targeted interventions in this complex syndrome.
Plain Language Summary
This review synthesizes recent evidence demonstrating that NRP1 integrates signals from multiple pronociceptive mediators enriched in the TME, including nerve growth factor (NGF), vascular endothelial growth factor A (VEGFA), hepatocyte growth factor (HGF), and platelet-derived growth factor (PDGF). We detail the function of NRP1 on amplifying nociceptive transduction. The review further examines how NRP1 contributes to three interconnected pathophysiological axes: the transduction of nociceptive stimuli, the induction of neuropathic changes, and the maintenance of inflammatory sensitization. We also address the role of NRP1 in therapy-related pain including chemotherapy-induced peripheral neuropathy, compensatory signaling during targeted therapy, and indirect pathway activation in immunotherapy. Finally, we evaluate the therapeutic rationale for targeting NRP1, both as a standalone strategy and in combination with conventional opioids, and discuss current translational challenges and future research priorities.
1. Introduction
Pain is a near-universal and profoundly debilitating consequence of advanced cancer, affecting up to 90% of patients with metastatic disease.1-3 Beyond the suffering it causes, pain erodes quality of life, limits functional capacity, and often undermines the tolerance and efficacy of curative cancer treatments. 4 The conceptual framework for understanding cancer pain has shifted. It is no longer viewed as a passive result of tumor compression, but rather as an active, dynamic process driven by the evolving biochemistry of the malignant niche.5-7
This process is sustained by three interconnected pathological drivers that together create a self-perpetuating nociceptive cycle: (1) continuous release of algogenic factors, including nerve growth factor (NGF), vascular endothelial growth factor A (VEGFA), and endothelins in the tumor microenvironment (TME); (2) iatrogenic biochemical perturbations from chemotherapy and radiation that directly injure neurons and remodel the tissue environment; and (3) the consequent establishment of a chronically inflamed TME characterized by immune infiltration, acidosis, and hypoxia. These elements converge on the peripheral and central nervous systems, inducing persistent peripheral sensitization (hyperexcitability of primary nociceptors) and central sensitization (maladaptive plasticity in spinal and supraspinal pain circuits).8-12 These mechanisms account for the chronic, severe, and often treatment-resistant nature of cancer pain.
Within this complex framework, molecules capable of integrating signals from multiple convergent pathways hold exceptional strategic value both for deciphering disease mechanisms and for identifying therapeutic targets. Neuropilin-1 (NRP1), a transmembrane glycoprotein initially recognized for its roles in angiogenesis and axon guidance, has recently emerged as precisely such a coordinator in cancer pain.13-15 NRP1 functions as a high-affinity co-receptor for several pronociceptive ligands enriched in the TME, including VEGFA, NGF, hepatocyte growth factor (HGF), and semaphorins. Although it lacks intrinsic kinase activity, NRP1 potently modulates signal transduction by assembling with the cognate receptors for these ligands (e.g., VEGFR2, TrkA, Met, plexins).16-20 This positioning enables NRP1 to act as a signal integration hub that amplifies and synchronizes inputs from diverse tumor- and therapy-driven pathways.
This narrative review dissects the molecular mechanisms by which NRP1 converts these multifaceted signals into sustained nociceptive neuronal activation. We summarize its tripartite role in: (1) initial nociceptive transduction through amplification of ligand–receptor interactions; (2) induction of neuropathic changes that establish chronic neuronal hyperexcitability; and (3) perpetuation of inflammatory sensitization within the TME, including its proposed involvement in the emerging “pain–immunosuppression” feedback loop. We further critically evaluate the translational promise of NRP1-targeted analgesia—both as a standalone strategy and as a rational complement to conventional opioid regimens—with the ultimate aim of intercepting the convergent pathways that sustain intractable cancer pain. This review is guided by the Scale for the Assessment of Narrative Review Articles (SANRA). 21
2. Discussion
2.1. NRP1: A Multifunctional Co-receptor in Physiology and Tumor Biology
NRP1 is a type I transmembrane glycoprotein encoded on chromosome 10, first identified as a receptor for the axonal guidance molecule semaphorin 3A (SEMA3A). Its structure features a large extracellular region organized into discrete a1/a2 and b1/b2 subdomains, connected via a single transmembrane helix to a short cytoplasmic tail of about 40 amino acids. The a1/a2 domains primarily engage semaphorin family ligands, whereas the b1/b2 domains are dedicated to binding VEGFA and other related secretory factors. A conserved PDZ-binding motif within the cytoplasmic tail is essential for recruiting scaffolding proteins and connecting to downstream signaling adaptors, enabling efficient signal transduction across the single-pass transmembrane.22-24
Functioning as a versatile co-receptor, NRP1 lacks enzymatic activity but critically modulates signaling by assembling complexes with primary receptors. These partnerships increase ligand-binding affinity and direct downstream signal flow. NRP1 serves as a signaling node, partnering with VEGFR2 for VEGFA, with TGFβR1 for transforming growth factor-beta (TGF-β), Met for HGF, PDGFR for platelet-derived growth factor (PDGF), TrkA for NGF, and plexin receptors for semaphorins (SEMAs). Meanwhile, these ligands, including VEGFA, HGF, and SEMA3C can also simultaneously activate the integrin signaling pathway through NRP1. Through these interactions, NRP1 contributions to essential biological processes and tumor progression, including angiogenesis, cell proliferation, cell migration and invasion, anti-apoptosis, drug resistance, and immune modulation (Figure 1).25-30 The multi-receptor role of Neuropilin-1 in physiology and tumor pathology. Neuropilin-1, as a co-receptor of VEGFA/VEGFR2, TGFB/TGFBR1, HGF/Met, PDGF/PDGFR, NGF/TrkA, SEMA3A/Plexin, and VEGFA, HGF, and SEMA3C/Integrins pathways, activates downstream MEK/ERK, PI3K/Akt, p38MAPK, FAK, SRC, and ROCK signaling pathways to drive angiogenesis, proliferation, migration and invasion, anti-apoptosis, drug resistance and immune modification functions, and also may be involved in the regulation process of cancer pain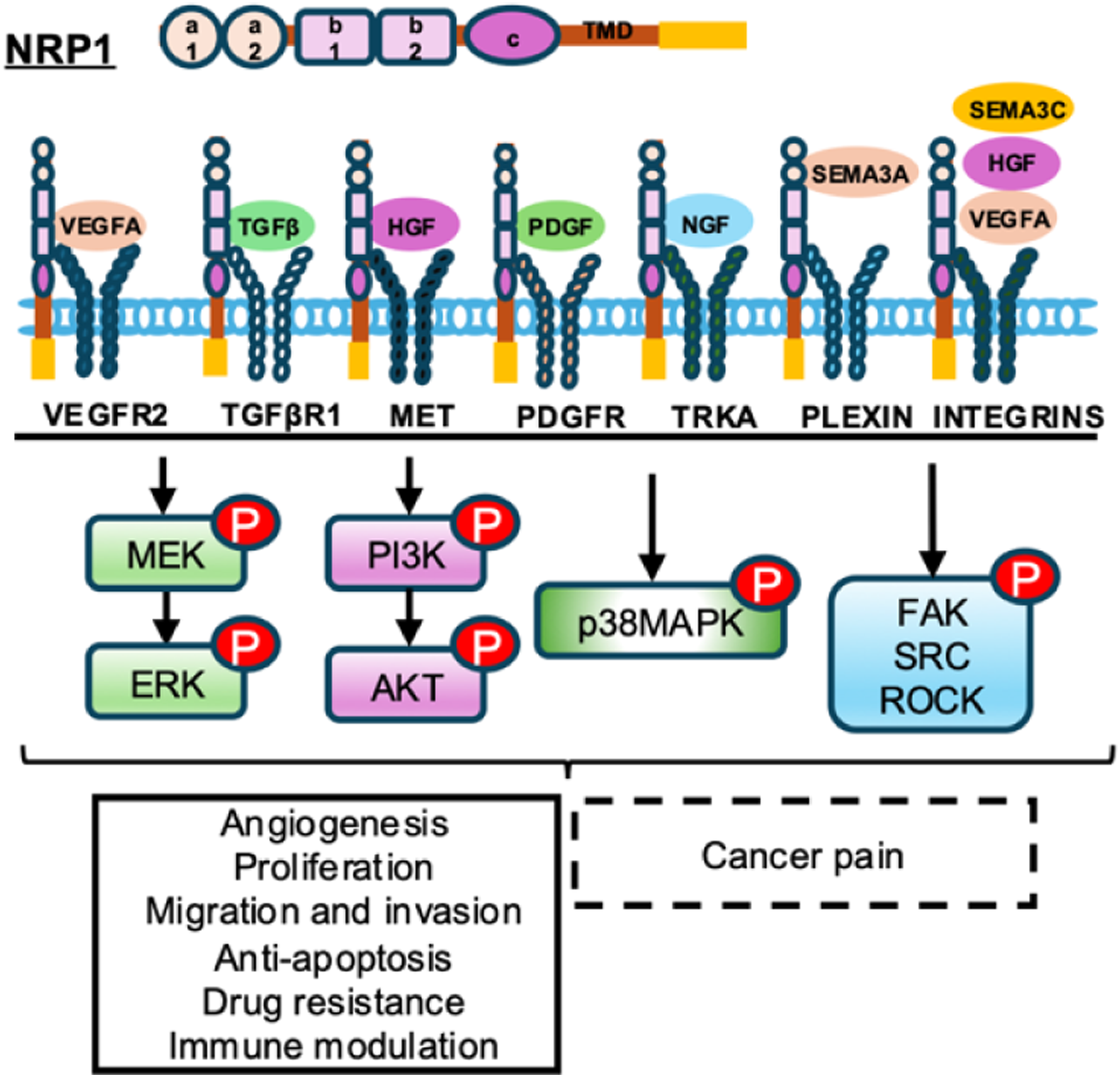
In cancer, NRP1 is hijacked to drive malignant progression. 3 31 A key mechanism is its pro-angiogenic function, where NRP1 acts as a co-receptor that amplifies signaling through the VEGFA-VEGFR2 axis. This promotes the formation of a dense but often aberrant tumor vasculature, which supplies oxygen and nutrients to support rapid tumor growth while also creating pathways for metastatic dissemination.32,33 Beyond angiogenesis, NRP1 directly regulates cancer cell behavior, fostering characteristic malignant traits such as uncontrolled proliferation, increased migratory potential, and tissue invasion. It plays a crucial role in initiating the epithelial-mesenchymal transition (EMT)—a developmental program exploited by tumors where cells shed adhesive properties, acquire motility, and develop apoptosis resistance, all critical for metastasis.34-36 Additionally, NRP1 contributes to therapy resistance, partly through compensatory activation of alternative receptor pathways that help cancer cells evade molecularly targeted drugs and chemotherapy.37-39
NRP1 also plays significant immunomodulatory roles within the TME. In regulatory T cells, NRP1 enhances stability, suppressive function, and tumor infiltration, thereby weakening antitumor immunity.40,41 Interactions with semaphorin ligands can alter dendritic cell maturation and T cell activation.42,43 By influencing vascular permeability and endothelial behavior, NRP1 further controls immune cell trafficking in tumors, helping to establish an immunosuppressive milieu that facilitates immune evasion.44,45
These pleiotropic effects are executed through the coordinated activation of key intracellular signaling hubs. NRP1-mediated signals converge on pathways such as PI3K/Akt, which strongly promotes cell survival and proliferation;46,47 p38 MAPK, involved in stress response and adaptation to the harsh tumor microenvironment;48,49 and FAK, SRC, and ROCK, the master regulators of cytoskeletal dynamics critical for cell adhesion, movement, and invasion.50,51 The integration of these signals underscores NRP1’s role as a molecular orchestrator of the aggressive cancer phenotype (Figure 1).
3. NRP1 as a Central Coordinator of Cancer Pain Signaling Within the Tumor Microenvironment
The NRP1 mediated nociceptive mechanisms that operate in cancer are not entirely unique to this disease. The downstream signaling pathways NRP1 engages appear to be shared across multiple chronic pain conditions. What distinguishes cancer is the concurrent surge of multiple NRP1 ligands such as NGF, VEGFA, and HGF, all released from a single expanding lesion. This simultaneous activation of parallel NRP1 dependent pathways may produce a greater degree of neuronal sensitization than that observed in inflammatory or neuropathic pain, where ligand elevation is often more limited. This cross condition relevance does not diminish the importance of NRP1 as a cancer pain target. On the contrary, it expands the therapeutic potential of NRP1 targeted agents. A drug that successfully blocks NRP1 driven pain in cancer patients might also relieve pain in other chronic pain syndromes characterized by elevated NRP1 ligands. The pathogenesis of cancer associated pain is multifactorial, arising from the interplay between direct tumor effects and dynamic host responses within the tumor microenvironment. This process can be understood through three interconnected pathophysiological mechanisms: transduction of nociceptive stimuli, induction of neuropathic changes, and maintenance of inflammatory sensitization. A unifying feature across these axes is the emerging role of NRP1. In cancer pain, far from being a passive bystander, NRP1 acts as a pivotal multifunctional co receptor and critical signaling hub. It integrates diverse cues from the tumor microenvironment, actively driving and amplifying each of these pathogenic pathways to establish and sustain the chronic pain state.
3.1. Transduction of Nociceptive Stimuli: From Ligand Binding to Sustained Signaling
NRP1 serves as a fundamental amplifier for the initial detection of tumor-derived algogenic signals. Its role is particularly well-defined within the NGF signaling pathway. Here, NRP1 is not merely ancillary but essential, binding NGF with high affinity (in the nanomolar range) to form a stable ternary complex with the TrkA, with a proposed stoichiometry of 2:2:2 (NGF:TrkA:NRP1). This interaction operates through a sophisticated dual mechanism: it concentrates local NGF for efficient presentation to TrkA, and acts as a chaperone to enhance TrkA trafficking from biosynthetic pathways to the plasma membrane, thereby increasing functional receptor density. The subsequent internalization of this complex, scaffolded by the adaptor protein Gα-interacting protein, C-terminus 1 (GIPC1) which links NRP1 and TrkA to the molecular motor myosin VI, is critical for generating sustained intracellular signaling from endosomes that powerfully excites nociceptors. 113,52 Similarly, within the VEGFA pathway, NRP1 potently enhances ligand binding and signaling through VEGFR2. This action not only contributes to indirect pain by promoting vascular permeability and edema,53,54 but more directly sensitizes pain-sensing neurons. VEGFA enhances currents through key voltage-gated sodium NaV1.7 and calcium CaV2.2 channels in dorsal root ganglion (DRG) neurons, lowering their activation threshold through the VEGFA/NRP1/VEGFR2 or VEGFA/NRP1 axis.14,15,55 By positioning itself at the apex of these key ligand-receptor systems, NRP1 effectively transforms transient chemical stimuli into a potent and prolonged nociceptive drive.
3.2. Induction of Neuropathic Changes: Architecting Neuronal Hyperexcitability
Moving beyond initial signal transduction, NRP1-propelled pathways are instrumental in driving the long-term maladaptive plasticity that characterizes neuropathic pain. Sustained VEGFA/NRP1/VEGFR2 signaling in sensory neurons promotes transcriptional and post-translational changes, leading to the upregulated expression and altered function of critical pain-related ion channels. 14 Genetic knockdown of NRP1 in DRG neurons using CRISPR/Cas9 editing has been shown to prevent VEGFA-mediated increases in CaV2.2 and NaV1.7 currents, directly linking NRP1 to this pathological ion channel remodeling. This results in lowered neuronal firing thresholds and increased spontaneous ectopic activity. Crucially, this pathogenic remodeling extends to the central nervous system.14,15NRP1-mediated signaling is implicated in enhancing excitatory synaptic transmission within the spinal cord dorsal horn—a core process underlying central sensitization. In vivo editing of NRP1 reduces the frequency of VEGFA-mediated increases in spontaneous excitatory postsynaptic currents in the spinal cord, demonstrating its role in facilitating pain signal amplification at the first central synapse.13,55 Thus, NRP1 evolves from a signal transducer to an architect of persistent peripheral and central neuronal hyperexcitability.
3.3. Maintenance of Inflammatory Sensitization: Integrating the Neuro-Immune Axis (Inferences)
The pro-nociceptive TME is defined by concurrently elevated cytokines, chemokines, and growth factors. Within this inflammatory landscape, NRP1 functions as a signal integrator rather than a passive relay. It simultaneously engages NGF, VEGFA, HGF, and PDGF, synergizing inputs that collectively sustain neuronal and glial activation.16,56,57 This signaling convergence feeds into a recently characterized “pain-immunosuppression” circuit. Activated nociceptors release Substance P and calcitonin gene-related peptide (CGRP). These neuropeptides do more than transmit pain—they suppress local antitumor immunity, inhibiting dendritic cell antigen presentation and impairing cytotoxic T lymphocyte recruitment.58-60 NRP1 reinforces this cycle through direct immunomodulation. It is expressed on a subset of Tregs and facilitates their infiltration into tumors in a VEGFA-dependent manner. 61 T cell-specific ablation of NRP1 in murine models reduces intratumoral Treg accumulation, restores CD8+ T cell effector function, and delays tumor growth without disrupting peripheral tolerance.62,63 The feedback loop is self-perpetuating. Tumor-derived factors drive NRP1-mediated nociception; the resulting neurogenic signaling fosters an immunosuppressive niche; tumor progression accelerates; pain intensifies. This interdependence positions NRP1 as a molecular bridge between chronic pain and immune evasion, with therapeutic blockade offering simultaneous analgesic and antitumor potential.
In summary, NRP1 orchestrates cancer pain through a tripartite and progressive mechanism. It initiates pain by potently amplifying acute nociceptive signals via defined ternary complexes, chronifies it by driving enduring neuropathic alterations in ion channel expression and synaptic function, and sustains it by integrating multiple inflammatory signals and linking nociception to tumor-promoting immunomodulation. This central, multi-mechanistic role elevates NRP1 from a simple co-receptor to a strategic therapeutic nexus (Figure 2). Pharmacological targeting of NRP1 holds unique potential to disrupt the cancer pain cascade at multiple points simultaneously, providing a compelling rationale for its development as a novel analgesic target for complex cancer pain syndromes. Neuropilin-1 regulates cancer pain through multiple comprehensive mechanisms. Cytokines including NGF, VEGFA, and HGF enter the circulation following secretion. VEGFA binding to NRP1 and VEGFR2 on endothelial cells drives vascular permeability and edema. This serves a dual function: edema generates mechanical pressure that activates nociceptors, while increased permeability facilitates cytokine transport to nerve terminals. At neuronal surfaces, these cytokines assemble into signaling complexes with NRP1 and their respective receptors. The NGF/NRP1/TrkA complex, stabilized by GIPC1, together with VEGFA/NRP1/VEGFR2 and HGF/NRP1/Met complexes, activate downstream kinase signaling to drive neuronal sensitization and pain transmission. Beyond direct effects on nociceptors, the same signaling events stimulate release of calcitonin gene-related peptide (CGRP) from sensory neurons. CGRP suppresses dendritic cell function and limits CD8+ T cell recruitment. This neurogenic suppression of adaptive immunity links tumor-driven pain directly to immune evasion
4. NRP1 Integrates Diverse Therapy-Induced Signals to Drive Persistent Cancer Pain
Beyond its well-established role in mediating pain directly driven by malignant tumors, NRP1 has emerged as a central molecular node in the pathogenesis of therapy-related cancer pain. Modern anticancer modalities—while indispensable for tumor control—each inadvertently remodel the tissue microenvironment in ways that converge upon NRP1 activation. Chemotherapy inflicts direct neurotoxicity, molecularly targeted agents provoke compensatory signaling rewiring, and immunotherapies induce systemic inflammatory cascades. Despite their distinct mechanisms of action, these diverse therapeutic insults share a common downstream consequence: the sustained engagement of NRP1 and its associated signaling networks, which translates acute treatment-induced injury into chronic, often refractory pain states that persist long after the offending therapy has been discontinued.
4.1. Chemotherapy: Direct Activation of NRP1-dependent Nociceptive Pathways
Cytotoxic chemotherapeutic agents, particularly microtubule-stabilizing agents such as paclitaxel and platinum-based compounds like oxaliplatin, represent the most extensively characterized iatrogenic cause of therapy-induced neuropathic pain, is also called chemotherapy induced peripheral neuropathy (CIPN). 64 These drugs exert direct, dose-limiting toxicity on peripheral sensory neurons, with DRG neurons being especially vulnerable. The initial insult involves mitochondrial dysfunction, oxidative stress, and the rapid release of damage-associated molecular patterns from injured neurons and surrounding satellite glial cells.65-67 This local injury response potently upregulates two key NRP1 ligands, VEGFA and NGF, within the DRG, the tumor microenvironment, and reactive glial populations.68-71 In the damaged peripheral nerves, the upregulation of histone H3 acetylation and trimethylation mediated in the VEGFA promoter region drives angiogenesis and persistent pain behavior. 69 Resident macrophages overexpress VEGFA to lower the mechanical pain threshold and open the blood-nerve barrier, allowing blood-derived cells and molecules to enter the nerve lining and induce neuropathic pain. 70 Cisplatin induces a neuroinflammatory environment in the DRG, driving CD11b and F4/80 positive macrophages to express more NGF, thereby enhancing nociceptive activity. 71 Subsequently, NRP1 acts as a central amplifier of these chemotherapy-induced injury signals: it forms the essential ternary complex with NGF and TrkA to enhance neuronal sensitization and co-receives VEGFA with VEGFR2 to potentiate the modulation of ion channels (NaV1.7 and CaV2.2) and neuronal hyperexcitability. Consequently, NRP1 may function as a central driver of chemotherapy-induced peripheral neuropath.
4.2. Molecularly Targeted Therapy: Compensatory and Ligand-Redundant Engagement of NRP1
Agents designed to intercept specific oncogenic driver pathways can inadvertently sustain pain signaling through NRP1 dependent compensatory mechanisms. Anti angiogenic therapy targeting the VEGFA/VEGFR2 axis, a cornerstone for many solid tumors, exemplifies this paradigm. Chronic VEGFR2 blockade often triggers adaptive transcriptional responses in the tumor microenvironment, including upregulated expression of alternative NRP1 ligands such as HGF and NGF, as well as increased NRP1 itself on tumor cells and stromal elements. Hormone therapy also provides a parallel example, although it effectively suppresses breast and prostate tumors, hormone therapy can also induce a rise in NRP1 expression, thereby driving treatment failure and tumor progression. In both scenarios, NRP1 integrates compensatory signals that sustain nociceptive pathways and malignant behavior.39,72-75 This compensatory upregulation has direct nociceptive consequences. The HGF/NRP1/Met and NGF/NRP1/TrkA axes, thus potentiated, actively drives neuronal sensitization. Concurrently, the sustained therapeutic pressure induces a state of ligand redundancy:76-78 VEGFA itself persists in the microenvironment and continues to drive pro-nociceptive signaling directly through the VEGFA/NRP1 axis, even when its canonical VEGFR2 pathway is effectively blocked. This mechanistic plasticity allows pain signaling to persist or even emerge de novo despite effective anti-tumor response, presenting a significant and under-recognized clinical challenge. Collectively, NRP1 integrates these aberrant compensatory and redundant signals, positioning itself as a common pathological effector downstream of diverse targeted therapy-induced adaptations.
4.3. Immunotherapy: Neuroinflammation and Indirect Pathway Activation (Inferences)
Immune checkpoint inhibitors (ICIs), which achieve anti-tumor efficacy by unleashing T-cell mediated immunity, can provoke a spectrum of immune-related adverse events, including neuroinflammation and inflammatory pain syndromes.79-81 The mechanistic linkage between ICI therapy and NRP1-mediated pain signaling is indirect yet compelling. The systemic and local release of pro-inflammatory cytokines, including VEGFA, tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), constitutes a hallmark of ICI-induced immune activation.82-85 These cytokines potently stimulate multiple cell types (including tumor cells, stromal fibroblasts, and infiltrating macrophages) to secrete VEGFA, NGF, and HGF.86-90
While direct evidence for ICI-driven transcriptional regulation of NRP1 remains an active area of investigation, the robust induction of its primary ligands provides a strong rationale for indirect pathway engagement. Furthermore, NRP1 itself is expressed on key immune effector populations, including Tregs and subsets of activated macrophages.91,92 Within the tumor microenvironment, NRP1 expression on Tregs reinforces their suppressive phenotype and promotes intratumoral infiltration in a VEGFA-dependent manner, contributing to an immunosuppressive niche.61,62 This raises the intriguing possibility of a bidirectional pain-immunosuppression loop: ICI-induced cytokine release drives VEGFA/NGF production, which activates NRP1 on both nociceptors (causing pain) and Tregs and macrophages (potentially dampening anti-tumor immunity). Thus, NRP1 may serve as a molecular bridge integrating immune-derived inflammatory signals with neuronal sensitization circuits, potentially exacerbating pain in a subset of ICI-treated patients.
In conclusion, NRP1 functions as a common pathological effector across the three major domains of modern anticancer therapy. Whether activated by the direct neurotoxicity of chemotherapy, the compensatory ligand plasticity induced by targeted agents, or the indirect cytokine-driven pathway engagement downstream of immunotherapy, NRP1 serves as a mechanistic bottleneck through which diverse iatrogenic insults are amplified and sustained as chronic pain. This ability of integrating signals from multiple, mechanistically distinct treatment modalities elevates NRP1 from a mere signaling co-receptor to a strategic therapeutic node (Figure 3). Pharmacological targeting of NRP1 offers the unique potential to uncouple this convergence, providing a rational, multi-indication strategy for the prevention and management of treatment-related pain across the oncologic care continuum. Neuropilin-1 mediates diverse therapy-induced signals to persist cancer pain. Chemotherapy drugs induce oxidative stress, mitochondrial damage, and inflammation. Molecularly targeted drugs activate compensatory and redundant signaling. Immunotherapy drugs lead to neuroinflammation and inflammatory factors release. These distinct mechanisms converge to upregulate the expression of NGF, VEGFA, HGF, and NRP1 within the tumor microenvironment, ultimately promote both tumor progression and cancer pain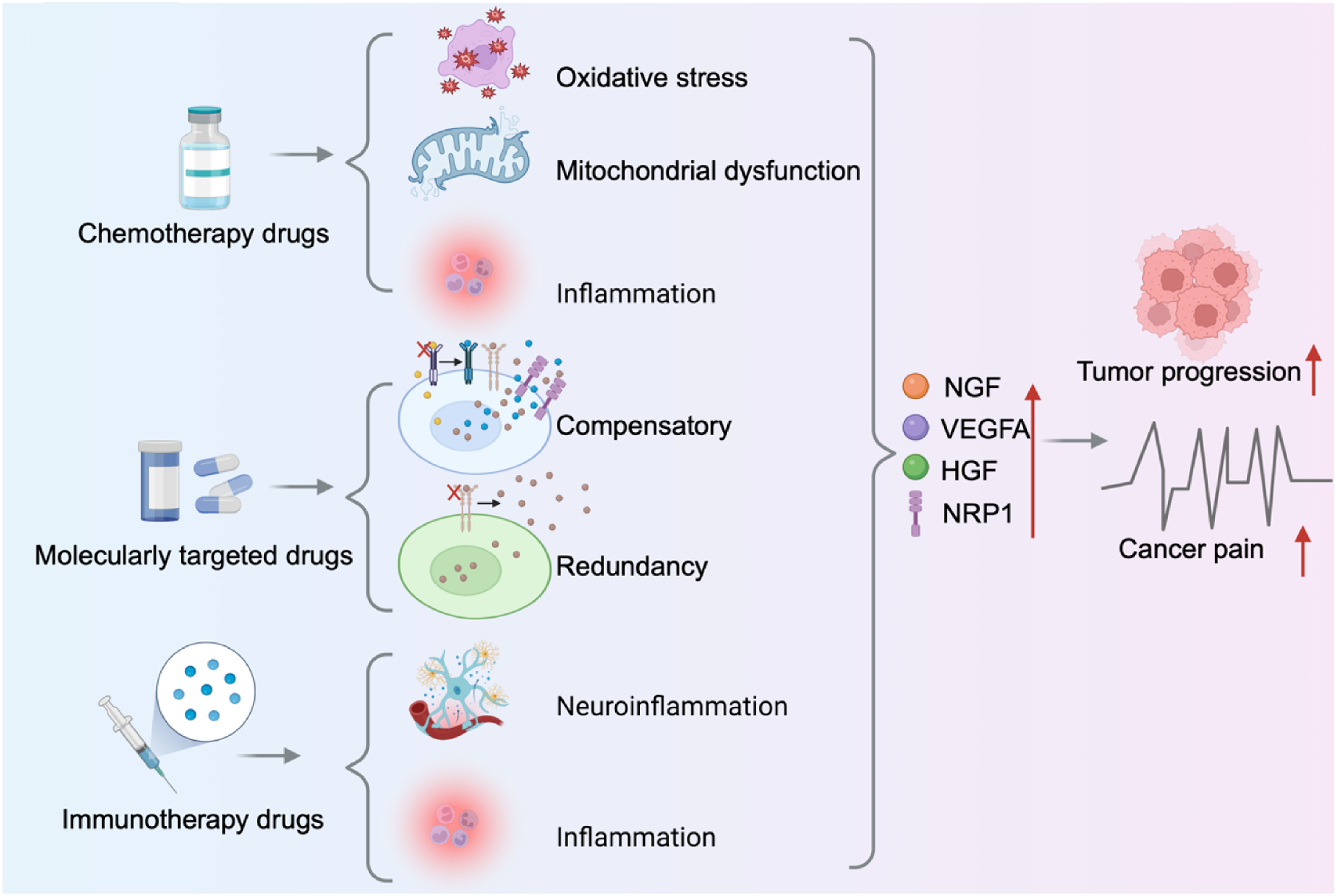
5. Therapeutic Targeting of NRP1 and Rationale for Combination Therapy
5.1. NRP1-Targeted Pharmacological Agents
The strategic inhibition of NRP1 represents a promising frontier in cancer pain management, with several distinct pharmacological classes demonstrating therapeutic potential. Current investigational agents can be divided into four platform classes: small-molecule antagonists, peptide-based agents, antisense oligonucleotides and monoclonal antibodies.
These four platform classes differ in their mechanisms and development status. Small molecule inhibitors include EG00229 and EG01449. The small-molecule inhibitor EG00229 attenuates NGF-induced thermal hyperalgesia in rodent models by selectively disrupting the NGF-NRP1 interaction. Similarly, intrathecal delivery of NRP1-targeting agents reduces pain behaviors in models of bone cancer pain and chemotherapy-induced neuropathy.13,15 Further evidence comes from the quinoline-based antagonist EG01449, which alleviates VEGFA-induced pain by modulating voltage-gated sodium and calcium channels in sensory neurons. 93 Peptide agents are represented by certepetide, also known as CEND1. This peptide binds NRP1 and αv integrins to activate the CendR pathway for tumour penetration. It has completed Phase II oncology trials, but pain specific endpoints have not been formally captured. 94 Antisense oligonucleotides, with SECN 15 as the lead candidate, downregulate NRP1 mRNA and protein expression and significantly enhance the anti-tumor efficacy of immunotherapy in preclinical models. 95 Monoclonal antibodies such as vesencumab (MNRP1685A) are human anti NRP1 antibodies that block ligand binding. A Phase I trial in advanced solid tumours has been completed, providing a clinical safety benchmark for this class. 96
Collectively, these findings validate NRP1 as a druggable target centrally involved in sustaining nociceptive signaling across diverse pain states, bridging direct analgesic effects with broader disease-modifying potential.
5.2. Unique Advantages of NRP1 among Established Cancer Pain Targets
Unlike NGF and TrkA, each of which defines a single ligand receptor pair, NRP1 works upstream as a shared co receptor for NGF, VEGFA, HGF, and PDGF. This positioning allows NRP1 to intercept multiple pronociceptive signals at the same time. Selective inhibitors of NGF or TrkA block only one pathway and come with notable side effects. The anti NGF antibody tanezumab caused rapidly progressive osteoarthritis and osteonecrosis in clinical trials.97,98 Similarly, the voltage gated sodium channel Nav1.7 shows a strong genetic link to pain insensitivity, yet its selective inhibitors have repeatedly failed in human trials due to species differences and functional redundancy among sodium channel isoforms. 99 NRP1 avoids these drawbacks by acting at an earlier, ligand convergent node. This may provide broader analgesic effects, and its restricted expression on nociceptors allows more precise targeting. Thus, compared with established cancer pain targets, NRP1 holds greater promise for clinical translation.
5.3. Rationale for Combination Therapy With Conventional Opioids
Convergent Principles of Combination of NRP1-Targeted Agents With Standard Opioid Regimens

MOR, mu-opioid receptor; NRP1, neuropilin-1; OIH, opioid induced hypersensitivity.
6. Challenges, Translational Hurdles, and Future Directions
NRP1 is now validated as a high-affinity co-receptor for NGF and a signal integrator for VEGFA, HGF, and PDGF in nociceptive neurons. This positions it as a rare, mechanism-based target for non-opioid cancer pain therapy. Translation, however, faces several distinct challenges.
6.1. Selectivity Versus Pleiotropy
NPR1 supports blood vessel formation, immune cell function, and vascular barrier integrity. A non selective, system wide blockade of NRP1 carries the risk of interfering with these essential physiological processes. The challenge is to interfere with NRP1 driven pain signaling without compromising its normal functions. The solved structure of the NGF–TrkA–NRP1 ternary complex and the identification of the C terminal NRP1 docking motif in NGF provide a blueprint. Targeting this interface could block NGF evoked nociception while preserving NRP1’s vascular and immune functions. Whether such molecular precision is achievable in humans remains the central open question.
6.2. Patient Heterogeneity and Biomarkers
NRP1-dependent pain is not universal. Its contribution varies by tumor type, stage, and treatment history. Prospective biomarker strategies are needed. Candidates include NRP1 expression in accessible tissues, circulating levels of NGF or VEGFA, and nociceptor-enriched NRP1 isoforms. Patients with high concentrations of serum NGF or VEGFA, or high expression of NRP1 in tumor tissues, may achieve better analgesic effects from NRP1 targeted drugs.
6.3. Pipeline and Lead Optimization
Several NRP1-targeted modalities are advancing. Peptides disrupting the NGF–NRP1 interface show efficacy in rodent pain models and human sensory neurons but require optimization of pharmacokinetics and stability. The small-molecule antagonist EG01449 blocks VEGFA-driven ion channel sensitization and reverses allodynia in preclinical models. The antisense oligonucleotide SECN-15, which knocks down NRP1 expression, enhances anti-PD-1 efficacy and is entering Phase I/II trials in oncology. 95 Whether it also suppresses pain signaling is an open, testable question.
6.4. Priority Research Agenda
(1) Selectivity. Develop inhibitors that discriminate pain-related NRP1 complexes (NGF/TrkA and VEGFA/VEGFR2) from those mediating angiogenesis or immunity. Allosteric small molecules, interface peptides, and conformation-selective biologics merit pursuit. (2) Combination regimens. NRP1 inhibitors are best positioned as opioid-sparing adjuvants. Rigorous preclinical studies should pair them with morphine, oxycodone, or fentanyl in validated cancer pain models (bone metastasis, CIPN). Endpoints must include opioid dose–response, tolerance, and hyperalgesia. (3) Biomarker-anchored trials. First-in-human studies should stratify patients by circulating NGF/VEGFA or tumor NRP1 expression, and incorporate quantitative sensory testing and patient-reported outcomes. Early trials should target populations with high NRP1 pathway relevance, such as NGF-high bone metastases or refractory CIPN. (4) Understudied ligands. HGF and PDGF-AA are established NRP1 ligands abundant in the TME, yet their nociceptive roles remain underexplored. Conditional knockouts and selective inhibitors are needed to assess whether HGF/NRP1/Met and PDGF/NRP1/PDGFRα axes drive pain in specific contexts. (5) Pain–immunity interface. NRP1 promotes both nociceptor sensitization and Treg-mediated immune suppression. If NRP1 inhibition relieves pain and enhances checkpoint blockade concurrently, it would offer a dual therapeutic advantage. Oncology trials of agents like SECN-15 should incorporate pain endpoints from the start.
In addition, there is no NRP1 targeted drugs have been approved for pain, and no clinical studies are currently underway. To accelerate development, three strategies can be pursued. First, repurpose existing tumor candidates such as the small molecule antagonists EG00229 or EG01449 to capture secondary pain endpoints. Second, use the Phase I/II safety data of the antisense oligonucleotide SECN-15 to expedite pain trials. Third, design dedicated proof-of-concept studies in patients with refractory bone metastases or chemotherapy induced peripheral neuropathy.
6.5. Concluding Perspective
The challenges are real but not unprecedented. The NRP1 field now possesses what many emerging targets lack: a structurally defined, human-validated mechanism; diverse, developable drug modalities; and a clear differentiation from the failed systemic anti-NGF approach. Whether these advantages translate into clinical analgesics depends on disciplined execution of the research agenda outlined above. The goal—effective, non-opioid, mechanism-based pain control for millions of cancer patients—justifies the effort.
7. Conclusion
NRP1 represents a master regulator of cancer pain by integrating signals from multiple pronociceptive mediators in the TME, including NGF, VEGFA, HGF, and PDGF. Targeting NRP1 offers a novel strategy to disrupt the core mechanisms driving pain sensitization. The combination of NRP1-targeted agents with traditional opioids holds significant promise for achieving superior analgesic outcomes through synergistic mechanisms, ultimately allowing for lower opioid doses, reduced side effects, and improved management of complex cancer pain. Blazing this new trail requires concerted effort to translate this compelling mechanistic rationale into clinical reality for patients.
Footnotes
Ethical Considerations
This manuscript is a review article and does not involve a research protocol requiring approval by the relevant institutional review board or ethics committee.
Author Contributions
SS, AL, NQ, and JA wrote the original draft. CL, ZX, and MY collected relevant materials. NQ and JA proposed the concept. NQ, GC, and JA revised the manuscript and supervised this study. The final version of the manuscript has been read and approved by all authors.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data are available from the corresponding author on reasonable request.
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
ChatGPT was used to support English language polishing during manuscript preparation. The authors subsequently reviewed and modified the content as needed and take full responsibility for the published work.