
Abstract
The salient neuropathological defect in fragile X syndrome is the overabundance of immature dendritic spines in cortical pyramidal neurons. This review examines this anatomical synaptic defect in the context of other alterations in synaptic and circuit plasticity in fragile X mice. In theory, abnormal spines could lead to dysfunctional circuits and vice versa, so it is still not clear which problem comes first. Because of the tight structure-function relationships at the synapse, and given the significant overlap between signaling pathways that regulate spine shape/dynamics and long-term synaptic plasticity (both of which involve proteins regulated by fragile X mental retardation protein [FMRP]), it is argued that the two defects cannot be separated. It will be critical to determine whether neurons that lack FMRP and demonstrate alterations in long-term potentiation/depression also fail to undergo the expected enlargement/shrinkage of dendritic spines associated with those forms of synaptic plasticity or to establish clear links from FMRP signaling to either spine instability or defective synaptic plasticity, especially during critical periods of brain development. The resulting data will be vital in guiding translational research that can identify novel molecular targets for therapy in this devastating disorder.
Keywords
Until recently, the pathophysiological mechanisms underlying most neurodevelopmental disorders characterized by autism and intellectual impairment remained a puzzle to scientists and clinicians alike. But this panorama is rapidly changing thanks to important advances in research on Rett syndrome, neurofibromatosis, tuberous sclerosis, and fragile X syndrome (FXS), in part because the genetic basis of these disorders has been identified. Among these, FXS has perhaps emerged as the prototypical disorder in which to study how altered signaling in certain molecular pathways leads to the synaptic defects and dysfunctional circuits that eventually cause learning disability, anxiety, or autistic traits. FXS is the most common single-gene cause of autism and mental impairment (Hagerman and others 2010), which has sparked great interest from the scientific community. A discrete but relatively subtle neuropathological abnormality has been identified in FXS: abnormal spines in the brain. This makes it easier to look for specific links between the gene defect and the synaptic alteration. In contrast, in neurofibromatosis and tuberous sclerosis, the anatomical defects are more widespread and include significant nonneuronal pathology and tumors that may have independent effects on circuit function. A genetic mouse model of FXS exists, in which the gene Fmr1, which codes for the fragile X mental retardation protein (FMRP), has been deleted (Dutch-Belgian Fragile X Consortium 1994). These Fmr1 knockout (KO) mice have abnormal social and anxiety-like behaviors, problems with learning and memory, and spine defects that are all reminiscent of what occurs in FXS. Drosophila and zebrafish models also exist (den Broeder and others 2009; Zhang and others 2001), and these have also contributed greatly to our understanding of the conserved roles of FMRP in neural development.
This review aims at examining closely the dendritic spine abnormality in FXS in the framework of functional deficits in brain circuits. Here, I consider whether the evidence supports a primary role for this postsynaptic defect in the pathophysiology of FXS or whether this pathological hallmark of FXS should be considered no more than an epiphenomenon of circuit dysfunction. For details on the epidemiology, genetics, and clinical features of FXS or on the complex role of FMRP in mRNA trafficking and local translation at the synapse, the reader is referred to several outstanding reviews on those topics (Bagni and Greenough 2005; Bassell and Warren 2008; D’Hulst and Kooy 2009; De Rubeis and Bagni 2010; Hagerman and others 2010; Penagarikano and others 2007).
Higher Spine Density in FXS: An Inconsistent Phenotype
For the past two decades, it has been assumed that the main neuropathological hallmark of FXS (and its mouse model) is that the density and length of dendritic spines in pyramidal neurons in the neocortex are abnormally high. However, the first reports of three autopsy cases of FXS did not find an elevated spine density and only noted the presence of “long, tortuous spines with prominent terminal heads and irregular dilations mixed in with normal short and stubby spines” (Hinton and others 1991; Rudelli and others 1985). The authors interpreted these findings as indicative of a delayed dendritic development and noted that similar findings had been reported in other types of mental impairment (Purpura 1974).
It was only later, when the Greenough lab examined dendritic spine morphology in cortical pyramidal neurons from three additional autopsy cases of adult individuals with FXS, that they found not only the prevalence of long and tortuous protrusions compared to controls but also a higher spine density (Irwin and others 2001). Over several studies, they also reported a higher than normal spine density in layer (L) 5 cortical pyramidal neurons from Fmr1 KO mice regardless of cortical region or mouse genetic strain (Comery and others 1997; Galvez and Greenough 2005; McKinney and others 2005), with only one exception (Irwin and others 2002) that found overall spine density to be normal. Because unusually long spines (but not higher spine density) had been previously reported in other types of mental impairment, it was proposed that the high spine density phenotype in FXS was something unique to this disorder (Irwin, Galvez, and Greenough 2000), and this led to the theory that the absence of FMRP might result in a failure to prune synapses (Bagni and Greenough 2005).
A careful assessment of all the major studies published by several independent groups on the topic of spines in Fmr1 KO mice reveals that the synaptic defect is actually quite variable, depending on the brain region being examined, the experimental methods used to stain and photograph neurons, or the age of the mouse (Table 1). In particular, several recent studies using live imaging should prompt us to reconsider the accepted dogma that there is an overabundance of spines in FXS. For instance, during early postnatal development of the neocortex in Fmr1 KO mice, the density of dendritic spines was reported to be normal in three recent studies, regardless of cortical layer (Cruz-Martin and others 2010; Harlow and others 2010; Meredith and others 2007). The only salient exception was a study that found a slightly higher spine density in the first postnatal week but not at two or three weeks of age (Nimchinsky and others 2001). Curiously, the same group did not see a higher spine density when they examined live neurons in organotypic slice cultures at the equivalent age.
Spine Alterations in Fmr1 KO Mice

BDA = biotinylated dextran; DIV = days in vitro; EM = electron microscopy; FITC = fluorescein isothiocyanate; GFP = green fluorescent protein; NA = not available.
Direct comparison for average spine length between Fmr1 knockout (KO) and wild type not available, but KO had more elongated thin protrusions and fewer shorter protrusions.
“Immature” in this table reflects the use of the term immature by authors or the observation of an overabundance of longer and/or more tortuous protrusions and fewer shorter or mushroom-like spines.
No retinal degeneration.
In contrast, in adult Fmr1 KO mice, Golgi studies in fixed tissue almost universally report a higher density of spines than in controls (Comery and others 1997; Dolen and others 2007; Hayashi and others 2007; Liu and others 2010; McKinney and others 2005), with few exceptions (Irwin and others 2002; Restivo and others 2005). This suggests that perhaps the spine density abnormality in FXS is an adult synaptic phenotype that is not seen during early development. This is unlikely, however, because a recent longitudinal in vivo two-photon imaging study reported that spine density is normal in juvenile and adult Fmr1 KO mice (Pan and others 2010). Instead, one must consider the possibility that the high spine density reported in adult Fmr1 KO mice (and in FXS autopsy cases; Irwin and others 2001) could perhaps be a histological artifact that is brought out by fixation and/or by the Golgi method. Why this would happen only to FXS tissue is hard to explain, but it may have to do with differences in spine lifetime (see below). It should also be noted, however, that a different Golgi study found that L4 neurons in Fmr1 KO mice have a normal spine density during the first postnatal week (Harlow and others 2010); this suggests that at least in early brain development, the Golgi method per se does not bring out a higher spine density in neurons from Fmr1 KO mice. In addition, differences in how analysis was performed by different groups could have led to contradictory results (e.g., using numbers of neurons instead of numbers of mice for statistics is likely to lead to more false-positive differences between mutant and wild-type [WT] mice).
Besides the effects of age and fixation/staining on spine density and length in FXS, attention has been drawn to the effects of several other variables on this synaptic phenotype, such as cortical layer, brain region, or mouse genetic background. Across cortical layers, results are mostly consistent, such that during early postnatal development, a normal density has been reported in excitatory neurons in L4 (Harlow and others 2010), L2/3 (Cruz-Martin and others 2010; Meredith and others 2007), and L5 (Nimchinsky and others 2001), although the latter study found a slightly higher spine density in the first postnatal week in fixed tissue but not in organotypic slices. Similarly, studies using adult fixed tissue consistently report a higher than normal spine density in mutant mice for pyramidal neurons in L5 (Irwin and others 2002; McKinney and others 2005) and L2/3 (Dolen and others 2007; Hayashi and others 2007). In contrast, across different brain regions, the available data show a wide range of spine phenotypes. For example, in the hippocampus, mutant mice have been reported to have either a lower spine density (Braun and Segal 2000; Segal and others 2003), which is the opposite of what is traditionally seen in the neocortex, or a normal spine density (de Vrij and others 2008; Grossman, Elisseou and others 2006; Pfeiffer and Huber 2007), regardless of age. Similarly, the length of spines on hippocampal neurons from Fmr1 KO mice has been reported to be either normal (Braun and Segal 2000) or increased (Bilousova and others 2009) compared to WT mice. Interestingly, even though genetic background is a major concern when looking at other FXS phenotypes, such as the susceptibility to audiogenic seizures (Yan and others 2005), the spine phenotype seems not to vary in any obvious pattern depending on the particular mouse strain. Thus, in the neocortex, a high density of spines has been seen in the c57Bl/6 and FVB strains of Fmr1 KO mice (Irwin and others 2002; McKinney and others 2005), whereas in the hippocampus, the differences in spine length in Fmr1 KO mice cannot be explained by the particular genetic background (Bilousova and others 2009; Braun and Segal 2000; see Table 1).
One concludes that spine abnormalities in fragile X mutant mice are consistent across cortical layers and genetic background and mostly consistent across different ages (as long as the staining method is the same), but they are quite variable across different brain regions. In particular, the discrepancy between the hippocampus and the neocortex is an important one because the altered mGluR plasticity (which has been demonstrated in the hippocampus but not in the neocortex) is at odds with the different spine defects reported in these two brain regions. Importantly, the most recent published data, including two live imaging studies of in the intact neocortex (Cruz-Martin and others 2010; Pan and others 2010), did not find a difference in spine density in Fmr1 KO mice. Accordingly, the field should perhaps abandon the idea that FXS is characterized by a failure to prune an excess of newly formed spine synapses. In fact, the opposite seems to happen—namely, that spines are abnormally unstable (see next section).
Considering that only six autopsy cases of FXS have been examined, the first three of which lacked thorough quantitative analysis, additional studies are required to clarify the exact abnormality of dendritic spines in humans. For example, it will be critical to analyze spines during early stages of cortical development in humans, before neurons begin to suffer from cumulative effects of seizures and other confounding diseases (or the medications used to treat them), and across different cortical layers. In addition, future studies of human autopsy material should report on spines in other regions besides the cortex that could give rise to symptoms in this disorder (e.g., hippocampus, cerebellum, amygdala) and examine autopsy cases of children of different ages if they become available. These studies should ideally be carried out with several different labeling techniques, such as filling of individual neurons with dyes such as Lucifer yellow (Elston and others 1997), to overcome the potentially confounding results of the Golgi staining method.
Immaturity and Instability of Spines in FXS
In contrast to the variable and sometimes contradictory findings regarding the density of spines in Fmr1 KO mice, what emerges as the most consistent abnormality from this retrospective look at the literature is the relative immaturity of spines. The notion that spines in FXS resemble dendritic protrusions during development dates back to the original human autopsy studies (Hinton and others 1991). A similar analogy also holds true for the Fmr1 KO mice, where a majority of published studies examining cortical neurons find an overabundance of thin filopodia-like spines and fewer mushroom-like spines, regardless of whether Golgi-stained cells are analyzed in fixed tissue (Comery and others 1997; Irwin and others 2002; McKinney and others 2005; Restivo and others 2005) or green fluorescent protein (GFP)–expressing neurons in the intact brain (Cruz-Martin and others 2010; Pan and others 2010). Furthermore, this immature spine phenotype is independent of whether or not a higher spine density is also found. Importantly, a similar overabundance of long, immature-appearing spines has also been reported in the hippocampus and in the cerebellum of Fmr1 KO mice (Antar and others 2006; Bilousova and others 2009; de Vrij and others 2008; Grossman, Elisseou and others 2006; Koekkoek and others 2005).
To arrive at the conclusion that spines are abnormally immature in this disorder, investigators originally used morphological criteria to distinguish between different categories of spines (Fig. 1). These classification schemes for spines in FXS (Irwin, Galvez & Greenough 2000) were inspired by static images obtained through electron microscopy (EM) (Harris and others 1992; Peters and Kaiserman-Abramof 1970). The assumption has always been that immature protrusions tend to be longer and lack bulbous swellings at the tips, whereas mature spines typically possess a large head at the tip, which is generally true in early development. But this classification has its critics (Bonhoeffer and Yuste 2002; Portera-Cailliau and others 2003) because it ignores the fact that spines and filopodia are highly motile in living neurons and can assume a variety of shapes and sizes over time scales of minutes to hours (Dailey and Smith 1996; Fischer and others 1998; Lendvai and others 2000; Portera-Cailliau and others 2003; Ziv and Smith 1996). This dynamic aspect of spines diminishes as neurons mature, but even in adult mice, spines can appear and disappear (or change shapes) over time scales of hours to days (Cruz-Martin and others 2010; Holtmaat and others 2005). Thus, a truly unbiased classification scheme for spines should rely not only on morphological characteristics (e.g., length, head width) but also on these dynamic features (e.g., motility, lifetime; Fig. 2). Preferably, it would distinguish protrusions on the basis of distinct molecular markers or on glutamate receptor content. However, to date, no specific markers that distinguish spines from filopodia have been identified, and documenting receptor content would require more complex experimental designs that include, for example, glutamate uncaging.
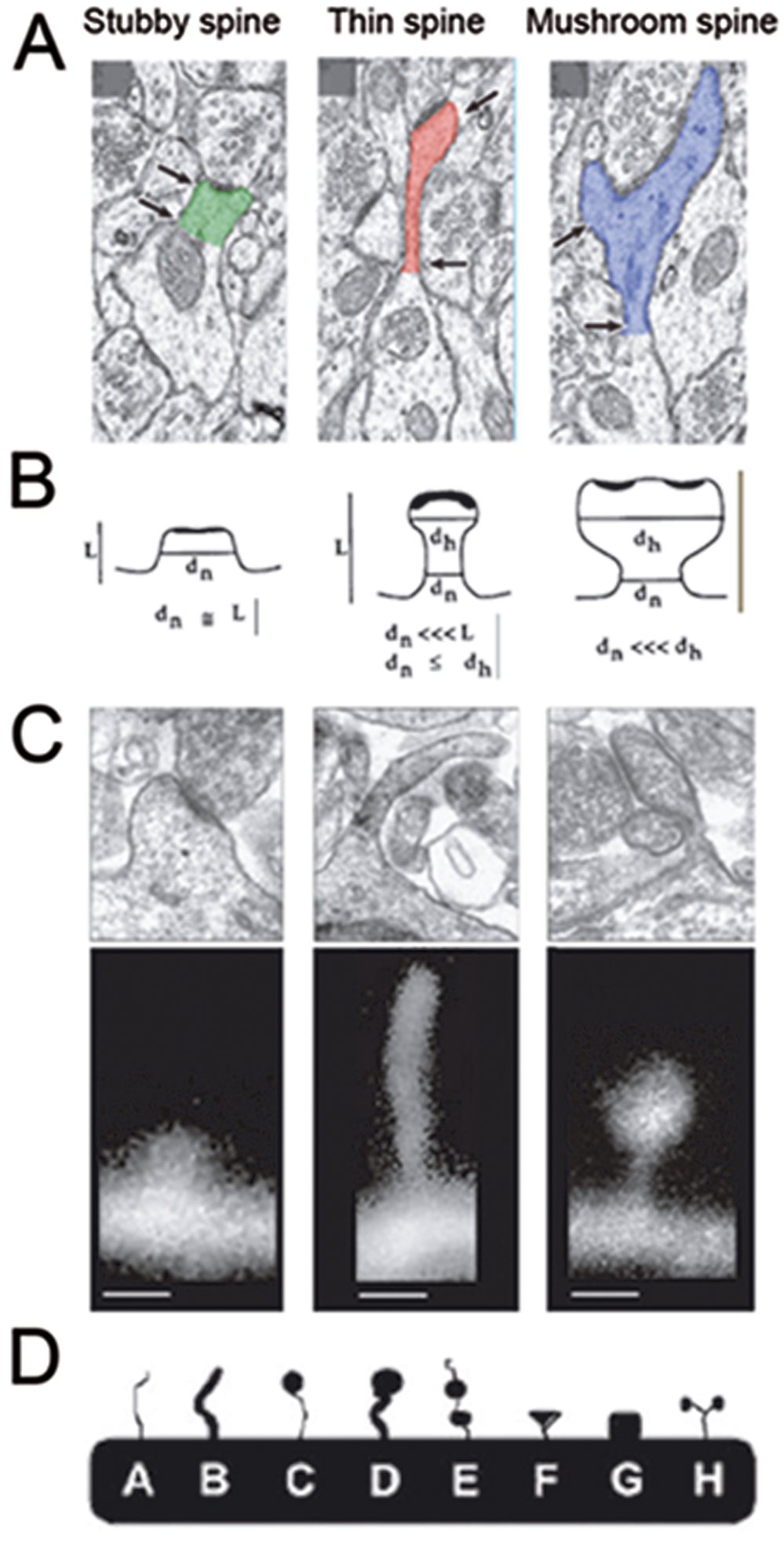
Traditional classification schemes for dendritic spines based on shape and size. (A) Electron microscopy studies have identified the existence of three different subclasses of mature spines: stubby spines, thin spines, and mushroom spines (Bourne and Harris 2008). (B) These ultrastructural studies suggested morphometric parameters to classify spines in these categories (Harris and others 1992). (C) Subsequent fluorescent imaging followed the same guidelines to categorize spines (De Roo and others 2008). A problem with this approach is that, when using static images, it is often difficult to distinguish between filopodia and thin spines or between short filopodia and stubby spines. (D) Other studies using the Golgi method for visualizing dendrites came up with slightly different classification schemes based on the shape of spines rather than the dimensions of the neck or head of spines (Grossman, Elisseou and others 2006). A problem with this approach is that it may only be applicable to Golgi-stained material.
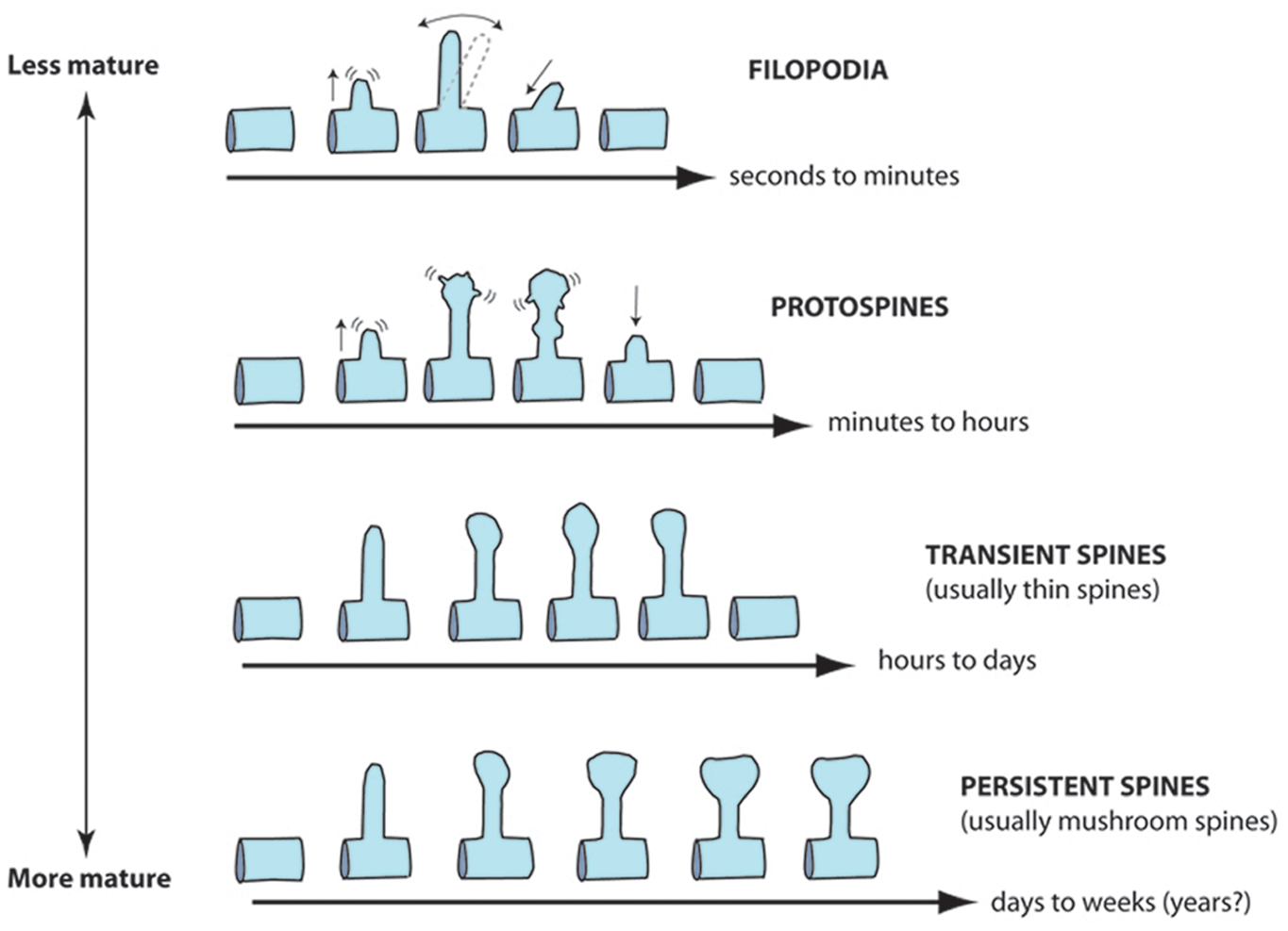
Proposed classification schemes for dendritic spines based on dynamics and lifetime. Because spines are motile structures whose shape and lifetime change throughout development, we propose a different classification scheme that incorporates these dynamic aspects of spines. We distinguish between filopodia (very short-lived protrusions that do not establish functional synapses), protospines (or spine precursors that establish rudimentary synapses early in development), transient spines (a mature protrusion that has a lifetime of less than a few days), and persistent spines (a mature protrusion that makes long-lived and stable synapses that last weeks to months, possibly years). Note that this classification scheme applies to pyramidal neurons in the neocortex and hippocampus. It is inspired by live imaging studies in slices and in vivo.
Recently, with the advent of two-photon microscopy and methods that allow the longitudinal imaging of fluorescent neurons in vivo through cranial windows (Mostany and Portera-Cailliau 2008), it has been possible to image dynamic phenomena of dendritic spines in their natural habitat (Holtmaat and others 2009). Using this approach, we imaged spines of L2/3 pyramidal neurons in the intact neocortex at various developmental stages (Fig. 3). We found that Fmr1 KO mice demonstrate a delay in the stabilization of dendritic spines because they exhibit an abnormally high turnover during the second postnatal week (Cruz-Martin and others 2010). This happens to correspond to the time when FMRP protein expression is highest in the cortex (Harlow and others 2010). Another group also showed an elevated spine turnover in L5 neurons of juvenile and adult Fmr1 KO mice when imaging in vivo over intervals of days and weeks (Pan and others 2010). Neither study could detect alterations in spine size or density, emphasizing how important it is to record the dynamics of spines in the intact brain. Next, it will be important to confirm that L2/3 neurons in juvenile mice also exhibit abnormally high spine turnover when imaged over longer intervals (days) than those we used to capture dynamics of the immature protrusions (one hour), but we suspect that the instability of spines in FXS is a long-lasting defect of pyramidal neurons across different cortical layers. One could speculate that strategies aimed at stabilizing dendritic protrusions might be of therapeutic value in FXS, particularly if implemented early in development. Candidate signaling pathways that are known to regulate spine size and/or turnover include the following: telencephalin (Matsuno and others 2006), cadherin/catenins (Abe and others 2004; Abu-Elneel and others 2008; Togashi and others 2002), neurotrophins (Luikart and others 2008), mTOR (Kumar and others 2005), Ephs/ephrins (Murai and others 2003), myosin IIb (Ryu and others 2006), and syndecan-2 (Ethell and Yamaguchi 1999). Ideally, future work should strive to adopt standardized techniques, analysis tools, and classification criteria. Whenever possible, investigators should use time-lapse imaging in vivo to remove the potential effects of disrupting brain circuits on spines (Kirov and others 1999) or to go beyond examining only static properties of spines. It is conceivable that fresh human tissue from FXS individuals might also become available (e.g., from epilepsy surgery), such that one might be able to examine spine motility and turnover in fluorescently labeled neurons.
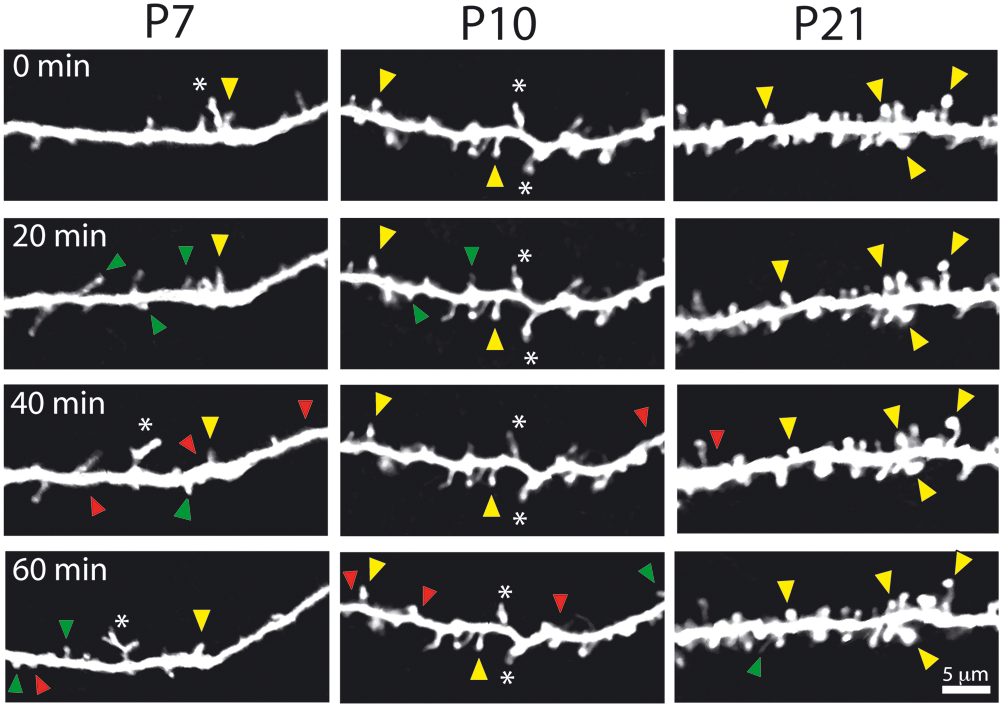
Abnormally high protrusion turnover in Fmr1 KO mice. Representative examples of time-lapse in vivo imaging of dendritic protrusions in Fmr1 KO mice at different postnatal ages. Images were collected every 10 minutes, but only half of the time points are shown for simplicity. Added, lost, and stable spines are labeled with green, red, and yellow arrowheads, respectively. Note also the presence of abnormally long and bright protrusions at early ages (asterisks). Reproduced from Cruz-Martin and others (2010) with permission from the Society for Neuroscience.
The failure of spines to stabilize during the critical period in barrel cortex strongly suggests that Fmr1 KO mice could have problems in maintaining the proper balance between stable and dynamic connections that is necessary to establish mature synapses. In the absence of FMRP, hippocampal neurons indeed have fewer spines that colocalize with synaptic markers, which suggests a loss of functional spines (Antar and others 2006). Surprisingly, little is known about the ultrastructure of these immature spine synapses. In their original article, Rudelli and others (1985) used EM to address this question in autopsy cases from FXS. They noted a marked reduction in the length of synapses and in the mean synaptic contact zone area, although other aspects of excitatory synapses appeared normal (synaptic vesicle density). Future work should examine spine synapses in Fmr1 KO mice at the subcellular level (e.g., synapse size, the presence of a spine apparatus, presence of polyribosomes, or the size of the postsynaptic density), perhaps through retrospective EM reconstructions of neurons previously imaged in vivo. Such studies are critical to establish the functional consequences of delayed stabilization of spines in FXS. For example, it is known that following LTP, the presence of polyribosomes correlates with a larger size of the postsynaptic density (PSD; Ostroff and others 2002), and yet polyribosomes are scarce in dendritic spines of Fmr1 KO mice (Weiler and others 2004). Considering the structure-function relationships at the synapse and in light of FMRP’s known role in regulating protein synthesis (see below), a compelling argument arises for how loss of FMRP might result in defects in activity-dependent regulation of spine morphology.
But why are spines slow to stabilize and mature in FXS? Speculating on this is a good exercise that generates a number of testable hypotheses (Fig. 4). In simplest terms, the main possibilities include either (1) a postsynaptic defect (e.g., alterations in scaffolding mechanisms that prevent spines from stabilizing or changing in response to appropriate afferent activity) or (2) a presynaptic defect (e.g., an abnormality in axon boutons or in afferent activity that makes them unsuitable to form stable synaptic contacts with dendritic protrusions). The rest of this review explores these possibilities in the context of different theories of FXS and what is known about FMRP signaling.
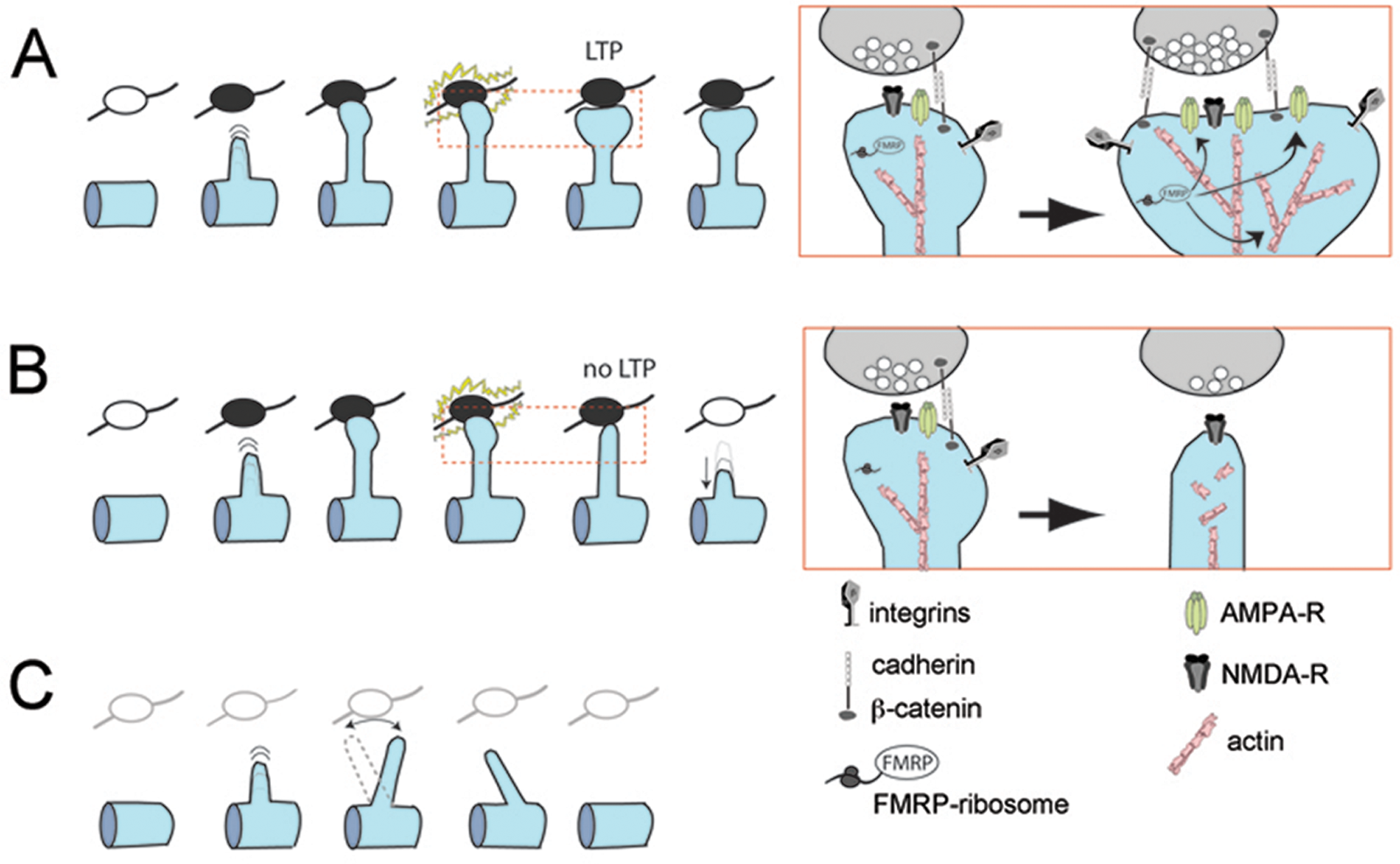
Possible causes of defect in spine stabilization in Fmr1 knockout (KO) mice: (A) Normal stabilization and enlargement of dendritic spines, with insertion of new AMPA receptor (AMPA-R) at the synapse, after long-term potentiation in wild-type mice. Insets in A and B show details of the effects of LTP on the actin cytoskeleton, AMPA-R trafficking, and the extracellular matrix scaffold (presumably mediated by proteins regulated by fragile X mental retardation protein [FMRP]). (B) Postsynaptic defect in Fmr1 KO mice: in the absence of FMRP, synaptic activity patterns that normally produce LTP fail to engage the actin cytoskeleton and scaffolding proteins needed to enlarge and stabilize the spine and simultaneously insert AMPA-R at the membrane. Ultimately, the spine is destabilized and retracts. (C) Presynaptic defect in Fmr1 KO mice: axonal boutons are either absent or unable to provide adequate afferent activity and fail to engage the postsynaptic spine into stabilizing.
Dendritic Spine Dysgenesis and Synaptic Plasticity Defects: Two Ways to Explain Symptoms in FXS
For two decades after the first description of aberrant spines in FXS, it was assumed that neuropsychiatric symptoms of affected individuals could be explained by the spine defect because their brains showed only negligible anatomical differences compared to age-matched controls (reviewed by Lightbody and Reiss 2009). Later, after the identification of the Fmr1 gene and FMRP, scientists began to look for links between FMRP signaling and the genesis/maintenance of dendritic spines. Not unlike other inherited forms of mental impairment characterized by immature-looking spines, which have been traced to genetic defects in scaffolding or adhesion molecules (Billuart and others 1998; Govek and others 2004; Newey and others 2005), FMRP was also quickly implicated in spinogenesis. The first direct connection emerged from work in Drosophila showing that FMRP binds to CYFIP1, which then binds to Rac1 (Schenck and others 2003). Interactions between FMRP and the Rac signaling pathway later emerged in mice too (Castets and others 2005). Rac1 is a member of the Ras superfamily of GTPases, which is known to regulate the actin cytoskeleton in all cells (Luo 2002). For example, neuronal overexpression of a constitutively active form of Rac1 results in a higher density of dendritic spines (Nakayama and others 2000; Tashiro and others 2000), whereas a dominant negative form of Rac1 results in a lower density of abnormally long spines (Tashiro and Yuste 2004). In addition, as an mRNA binding protein, FMRP directly regulates protein synthesis within dendritic spines (Bassell and Warren 2008), although it remains unclear which of these FMRP targets are involved in regulating spine formation, size, or dynamics. Among these many molecules, PSD-95 and CaMKII stand out because of their known effects on spine morphogenesis and synaptic strength (Grossman, Aldridge and others 2006; Muddashetty and others 2007; Zalfa and others 2007; Zalfa and others 2003). PSD-95 is particularly attractive because the activity-dependent stabilization of nascent spines is correlated with increased PSD-95 (De Roo and others 2008). FMRP also regulates two important scaffolding proteins at the PSD, Shank and SAPAP (Schutt and others 2009). It will be interesting to see whether loss of FMRP-mediated expression of such PSD proteins is responsible for the delayed stabilization of spines observed in Fmr1 KO mice. Last but not least, PI3K and PIKE, which are upstream of mTOR signaling and link FMRP more broadly to synaptic protein synthesis, could also be involved (Gross and others 2010; Sharma and others 2010). Overall, it makes sense that the absence of FMRP would directly affect spine morphology or dynamics via dysregulated protein synthesis, and a defect in the maturation of dendritic spines, the main recipients of excitatory synaptic inputs in the brain, would certainly explain deficits in intellectual ability seen in individuals with FXS.
But such a “spinocentric” view would ignore another equally important aspect of FXS research, which includes all the electrophysiology work that has demonstrated synaptic and circuit defects in Fmr1 KO mice. Motivated by the finding that Fmr1 KO mice exhibit deficits in spatial learning, which is associated with impaired LTP, initial studies examined hippocampal LTP in the mutant mice but failed to detect any deficits (Godfraind and others 1996; Paradee and others 1999). Shortly thereafter, however, a seminal study reported that mGluR5-dependent long-term depression (LTD) is exaggerated in Fmr1 KO mice (Huber and others 2002). In the resulting mGluR theory of FXS (Bear and others 2004), the primary defect in FXS was shifted away from a structural problem in spines and was linked instead to a functional deficiency in circuit plasticity. According to this view, the intellectual deficits of affected individuals, their propensity for seizures or obsessive-compulsive behaviors, even the immature dendritic spines, would all be explained by altered signaling through mGluR5.
Since the mGluR theory was first introduced, additional defects in synaptic plasticity have been uncovered in Fmr1 KO mice (reviewed in Pfeiffer and Huber 2009), including marked reductions in neocortical LTP (Desai and others 2006; Li and others 2002; Wilson and Cox 2007; Zhao and others 2005), impairments in spike timing–dependent plasticity in developing neocortex (Meredith and others 2007), and a general defect in Gq-coupled receptor-dependent LTD in the hippocampus (Volk and others 2007). What’s more, some of these abnormal synaptic plasticity phenotypes are being traced back directly to the absence of FMRP through established signaling pathways (Sharma and others 2010). Any of these alterations in synaptic physiology could easily explain the cognitive and behavioral symptoms of individuals with FXS. Consequently, one wonders whether deficiencies in LTP and LTD resulting from the absence of FMRP could lead to changes in the shape, size, or number of dendritic spines or whether instead they are a consequence of the aberrant spines in FXS. It becomes “the chicken or the egg question” of FXS: which comes first, dendritic spine dysgenesis or defects in circuit plasticity? To answer this crucial question, one must consider how spine size and maturity are related to synaptic strength and circuit plasticity.
Structure-Function Relationships at the Synapse and What This Means for FXS Research
Because dendritic spines lie opposite presynaptic boutons in excitatory synapses, a very tight structure-function relationship exists between spines and three aspects of neuronal activity: synaptic strength, synaptic forms of plasticity involved in learning and memory (LTP and LTD), and experience-dependent plasticity. The size of a spine is strongly correlated with the number of AMPA-type glutamate receptors and with the magnitude of excitatory postsynaptic potentials (reviewed in Kasai and others 2010). Thus, small filopodia-like spines tend to lack AMPA receptor (AMPA-R) currents, whereas larger mushroom spines have large AMPA-R currents (Matsuzaki and others 2001; Zito and others 2009). When we compared the average volume of all dendritic protrusions in Fmr1 KO mice and WT mice, we detected no differences at P10-12 (Cruz-Martin and others 2010), which is when they were abnormally unstable. Even in adults, spine volume is normal in Fmr1 KO mice (Pan and others 2010). In addition, because newly formed spines tend to be smaller (Holtmaat and others 2006), one might think that spine lifetime might correlate with synaptic strength. However, the amplitude of AMPA-R currents for a given spine is generally independent of its lifetime (Zito and others 2009). Therefore, the immaturity of dendritic spines in FXS does not necessarily mean that they establish weaker synapses; indeed, this is consistent with the normal amplitude of spontaneous miniature excitatory postsynaptic currents (EPSCs) in these animals (Desai and others 2006).
Strikingly, the formation and maturation of spines during development seem to proceed concurrently with the expression of functional AMPA-R currents, and the process is rather short (Zito and others 2009). Thus, under normal conditions, the majority of spines, even those with lifetimes of only tens of minutes during development, can establish functional synapses. In the adult cortex, spine growth usually precedes synapse formation (Knott and others 2006), which implies that the scaffold machinery needed to generate a spine can be assembled in the absence of functional glutamate receptors. From this information, we can surmise two things. First, that during early postnatal development, the short lifetime of spines in mutant mice might be related to an inability to establish synaptic contacts (as either the cause or the consequence). This would not be true of the elevated turnover in adults (Pan and others 2010) because spine lifetimes remain relatively long, which should still provide ample time to make a synapse. Second, a shorter than normal lifetime at any age might pose a problem not for establishing synapses but for the proper functioning of synapses, for example, during learning or experience-dependent plasticity.
Given the strong correlation between synaptic strength and spine volume, it is not surprising that dendritic spines would also be intimately associated with forms of synaptic plasticity involved in learning and memory. LTP and LTD are associated with the enlargement and shrinkage, respectively, of individual dendritic spines (reviewed by Kasai and others 2010). The functional readout of such changes in spine structure is mediated by glutamate receptor trafficking at the synapse, with AMPA-Rs being inserted (or removed) from the plasma membrane during LTP (or LTD) to modify synaptic strength. Here again it is difficult to tease apart which happens first, and most likely the processes of LTP, receptor trafficking, and persistent spine changes are tightly coupled and happen nearly simultaneously. For example, it is clear that LTP cannot occur with just a transient enlargement of the spine head—the spine growth must be maintained—and that AMPA-R trafficking alone (without spine enlargement) is not sufficient to ensure long-term changes in synaptic strength (Kasai and others 2010).
Protein synthesis at the synapse is required to implement not only the long-lasting changes in synaptic strength but also the persistent spine enlargement necessary for the establishment and maintenance of LTP and LTD (Kasai and others 2010). Of course, protein synthesis happens to be regulated by FMRP (Bassell and Warren 2008; D’Hulst and Kooy 2009; Penagarikano and others 2007), so it follows that the absence of FMRP would negatively affect synaptic plasticity and impair normal learning and memory. In fact, synaptic delivery of GluR1-containing AMPA-R, which is protein synthesis dependent (Sutton and others 2006), is impaired in cortical neurons of Fmr1 KO mice, and this probably contributes to the loss of GluR1-dependent LTP in the mutant mice (Hu and others 2008). The critical experiment that has not yet been done would be to look for specific defects in spine size during LTP/LTD in Fmr1 KO mice; one predicts that neurons from the mutant mice will be unable to exhibit structural plasticity. These experiments could be done in a mosaic setting, where only a minority of the neurons lacks FMRP, to minimize presynaptic effects that could potentially confound the results (see below).
Structural dendritic spine plasticity is also intimately associated with experience-dependent plasticity, such that sensory deprivation triggers changes in the turnover or motility of spines in the neocortex (reviewed by Holtmaat and Svoboda 2009). Plucking whiskers is associated with a significant increase in the motility of immature dendritic protrusions in barrel cortex at P9-P11 (Lendvai and others 2000), which is the same age when we observed a transient defect in protrusion dynamics in Fmr1 KO mice (Cruz-Martin and others 2010). Circuit rewiring after sensory deprivation does not affect all spines indiscriminately. For example, trimming of whiskers on the snout specifically stabilizes new spines but destabilizes previously persistent spines in barrel cortex (Holtmaat and others 2006; Wilbrecht and others 2010). This is important because persistent spines (defined as those that last weeks) tend to be much larger than transient spines (Holtmaat and others 2006). This means that changes in sensory experience affect synaptic strength through changes in dendritic spines. Because children with FXS experience hypersensitivity to sensory stimuli, one wonders whether those symptoms reflect problems with experience-dependent circuit rewiring during development at the level of spines. These issues have been examined in Fmr1 KO mice, and they clearly exhibit deficits in some forms of experience-dependent plasticity (Bureau and others 2008; Dolen and others 2007). Furthermore, sensory deprivation in adult Fmr1 KO mice fails to modulate dendritic spines (Pan and others 2010) or circuits in the cortex (Bureau and others 2008) the way it does in wild-type mice. The link between spines and altered sensory experience may occur very early in development, during specific critical periods. For example, Fmr1 KO mice show altered maturation of glutamate receptor signaling at thalamocortical synapses as early as P4-P7, which is probably the earliest known defect of any kind in Fmr1 KO mice (Harlow and others 2010). Considering that L4 matures before L2/3, it would be intriguing to find out if the same delay in spine stabilization that we report in L2/3 at P10-12 occurs earlier in L4.
In summary, the structural properties of spines (e.g., total volume, PSD size) cannot be separated from the functional or electrochemical properties of spines (e.g., AMPA-R currents). In fact, this relationship validates the two main phenotypes in FXS: the abnormal spines and the defects in circuit plasticity.
Spine Defects in Fmr1 KO Mice, mGluRs, and Synaptic Plasticity
Few discoveries, if any, have fueled as much interest and excitement in the past decade of FXS research as the finding of enhanced mGluR-dependent LTD in the hippocampus of Fmr1 KO mice (Huber and others 2002). The mGluR theory of FXS (Bear and others 2004) propelled the field forward by expanding the horizon of basic research into novel signaling pathways and by opening new avenues for therapeutic intervention in this devastating disorder. According to the original hypothesis, the way that enhanced mGluR-dependent LTD in hippocampal synapses might bring out the spine phenotype in FXS is via the internalization of AMPA-type ionotropic glutamate receptors, which reverts spines to a smaller size that is reminiscent of immature protrusions. In fact, the mGluR agonist 3,5 dihydroxyphenylglycine (DHPG) can elongate dendritic spines (Abu-Elneel and others 2008; Vanderklish and Edelman 2002). Also, Fmr1 KO mice with a 50% reduction in mGluR5 expression no longer have a high spine density (Dolen and others 2007), although it is not clear if the immaturity of spines was also reversed, as predicted by the mGluR theory. But other lines of evidence question such a link between mGluR signaling and spines. One conflicting point is that altered mGluR5 plasticity has only been reported in the hippocampus, where spine density is either normal or reduced (see Table 1). Until similar defects in mGluR-dependent LTD are revealed in the neocortex of mutant mice, it will be difficult to reconcile this discrepancy. Similarly, because mGluR-dependent LTP is absent in the neocortex of Fmr1 KO mice (Wilson and Cox 2007), it becomes challenging to explain how loss of FMRP triggers exaggerated mGluR signaling in some brain regions but not in others. The fact that the synaptic mechanisms of hippocampal mGluR-dependent LTD switch during early postnatal development (Nosyreva and Huber 2005) adds another layer of complexity to the interpretation of structural and functional alterations at the synapse. A better understanding of how FMRP regulates key proteins at the synapse during LTP and LTD across various brain regions and developmental stages will no doubt shed light on these issues.
In contrast, when one considers the impact of the functional defect (excess LTD in hippocampus vs. absent LTP in cortex) on the synaptic phenotype (depotentiation of synapses → loss of AMPA-R and immature spines) together, the results are consistent across brain regions. This supports a view of FXS in which the final readout of synaptic strength (spine volume and AMPA-R content) is similarly compromised in the neocortex and hippocampus, but the common link is not so much mGluRs but rather the absence of FMRP-dependent protein synthesis that regulates the actin cytoskeleton and scaffolding at the PSD, which ultimately dictates spine shape, receptor trafficking, and sustained changes in synaptic strength. For example, the absence of FMRP can be associated with low levels of PSD-95 (Zalfa and others 2007) or with failure to synthesize PSD-95 upon stimulation (Muddashetty and others 2007; Todd and others 2003). Because PSD-95 levels are important determinants of AMPA-R content at synapses (Colledge and others 2003), this could explain why Fmr1 KO mice have defects in N-methyl-
Other Confounding Issues: Presynaptic and GABAA Dysfunction in FXS
FXS has traditionally been thought of as a disorder of the postsynaptic compartment, presumably because of the spine anomaly and the fact that FMRP is concentrated in spines. More recently, however, several different lines of evidence point to a presynaptic role for FMRP, and the idea that at least some symptoms in FXS could be due to presynaptic dysfunction has been proposed. First, FMRP has been shown to be present in presynaptic terminals, where it exists in discrete granules in vitro (Antar and others 2006) and in vivo (Christie and others 2009). The expression of such presynaptic FMRP granules is regulated both developmentally and regionally in the brain, being maximal in the frontal cortex and hippocampal area CA3 in two-week-old mice but virtually nonexistent in adult neocortex or in CA1 (Christie and others 2009). In a different study examining mouse barrel cortex, postsynaptic FMRP expression was detected diffusely and at all ages, peaking at two to three weeks of age, whereas presynaptic FMRP was seen at P14 and P35 but not at P7 (Harlow and others 2010; Peter Kind, The University of Edinburgh, Scotland, 2010, personal communication). A second line of evidence comes from Drosophila, where mutations in the Fmr1 gene homologue result in defects in axon targeting and pruning (Tessier and Broadie 2008; Zhang and others 2001). Perhaps consistent with this finding, in Fmr1 KO mice, the L4 → L3 axonal projection in barrel cortex is more spatially diffuse than in wild-type controls (Bureau and others 2008). In addition, the strength of that ascending cortical projection was reduced in two-week-old mutant mice but not at later ages, which corresponds to the critical period for that projection in barrel cortex and happens to coincide with the developmental time period when spines of L2/3 neurons are too unstable (Cruz-Martin and others 2010).
Overall, this evidence for presynaptic defects in FXS is intriguing, but the relatively delayed and region-specific expression of FMRP in boutons compared to spines cannot easily explain the instability of spines in both developing and adult neocortex (Cruz-Martin and others 2010; Pan and others 2010) or in hippocampal area CA1 (Bilousova and others 2009). More work will be needed to elucidate the relationship between presynaptic FMRP granules and postsynaptic alterations in structural and functional synaptic plasticity. It will be equally important to further delineate postsynaptic defects in axonal structure and dynamics, preferably following an in vivo imaging approach (De Paola and others 2006; Portera-Cailliau and others 2005). In addition, studies in animals with mosaic Fmr1 gene expression will be crucial in teasing apart pre- and postsynaptic effects of FMRP (Hanson and Madison 2007).
The fact that a significant proportion of individuals with FXS experience hypersensitivity to sensory stimuli and seizures (Hagerman and others 2010) has led to the proposal that brain circuits may be hyperexcitable in this disorder. This question has been addressed from the point of view of inhibition in Fmr1 KO mice, and several lines of evidence now suggest that defects in inhibition could contribute to these symptoms (Reviewed by D’Hulst and Kooy 2007). Studies have shown a marked decrease in the density of parvalbumin-expressing interneurons in L4 (Selby and others 2007), a lower expression of the β and δ subunits of the inhibitory γ-aminobutyric acid (GABA) GABAA receptor at the protein level (Curia and others 2009; El Idrissi and others 2005), or a reduction in mRNAs coding for several GABAA receptor subunits in both mutant mice and flies, especially the δ subunit (D’Hulst and others 2006). Interestingly, the δ subunit, which together with α5 subunits mediates tonic inhibition (Glykys and others 2008), is an FMRP ligand (Miyashiro and others 2003), and Fmr1 KO mice have diminished tonic (but not phasic) inhibition (Curia and others 2009). In another study, a 50% reduction in excitatory drive onto fast-spiking interneurons (“feedback inhibition”) was identified in Fmr1 KO mice, whereas “feedforward inhibition” was found to be intact (Gibson and others 2008). Overall, the evidence is fairly compelling that inhibitory dysfunction is a real problem in FXS. But whether this is directly related to the absence of FMRP or is an end result of circuit dysfunction in glutamatergic circuits remains to be seen. Besides, a clear link between such defects in GABAA signaling and the abnormal dendritic spine phenotype has not yet emerged. The effect of GABAergic drugs on dendritic spines has not been thoroughly investigated, although one report found that benzodiazepines interfere with normal spine maturation (Tan and others 2009), which is the opposite of what the GABAA theory of FXS would predict. Further investigation of the effects of acute or chronic manipulations of various GABA receptor subunits on the morphology and dynamics of dendritic spines of pyramidal neurons should shed light on this issue, especially when one considers the important role of inhibition in critical period plasticity (Hensch 2005). One also anticipates that clinical trials will soon begin to assess the potential benefit of GABAergic drugs in patients with FXS.
Summary
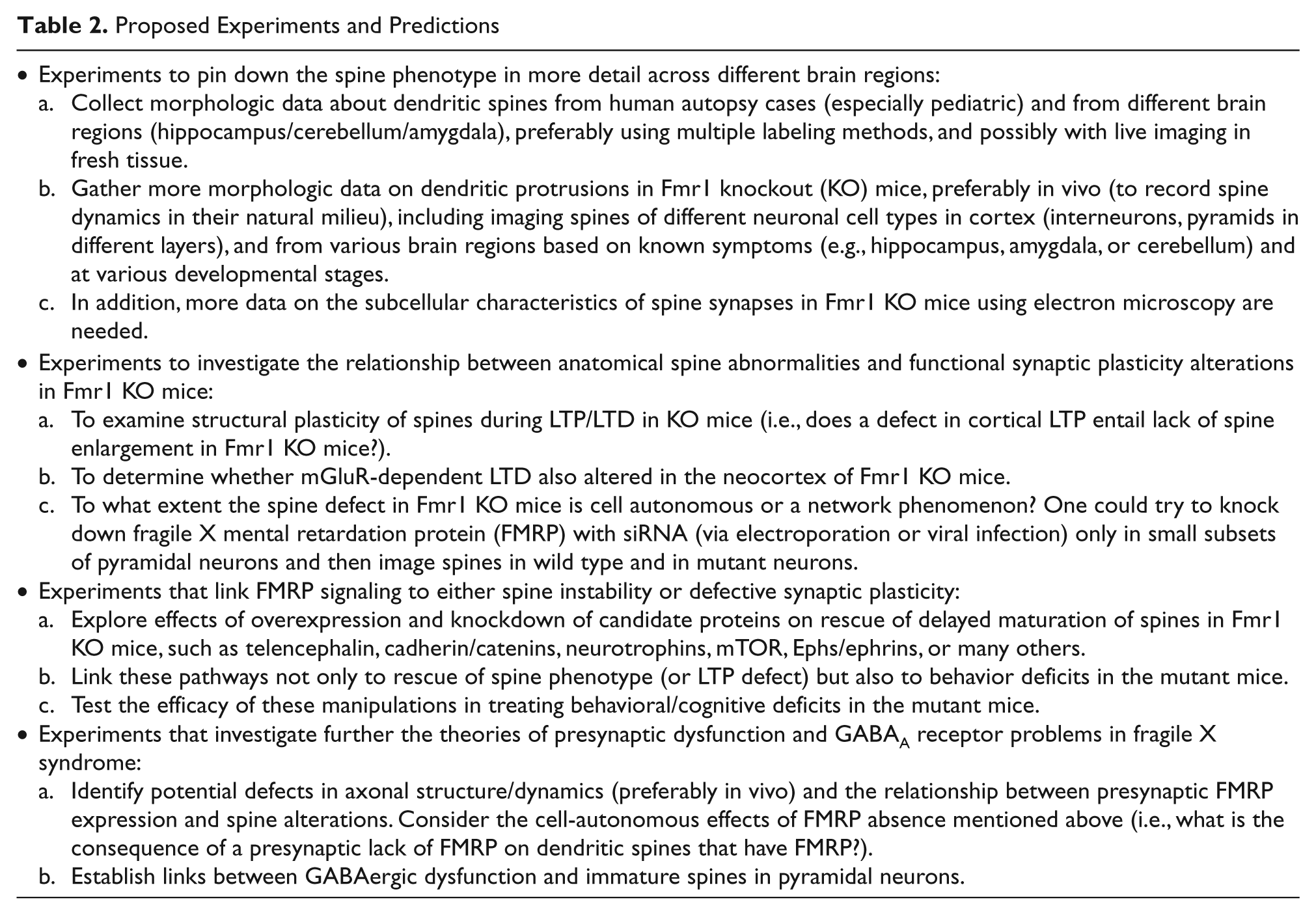
A better understanding of the cause-and-effect relationship between the well-known abnormalities in synaptic plasticity and dendritic spines in FXS will require additional experiments (Table 2). Based on what we know about synaptic strength, glutamate receptors at the synapse, and spine size and maturity, it is likely that the two problems are intimately linked to one another. Therefore, the more we study the signaling pathways that regulate the formation, size, and lifetime of dendritic spines, the more we will know about the alterations in functional synaptic plasticity in Fmr1 KO mice. Importantly, many of these defects in spines and in synaptic/circuit plasticity occur during critical periods of the neocortex in the first postnatal weeks, which coincides with the maximal expression of FMRP. Much of the available evidence points to FMRP playing a crucial role in experience-dependent circuit rewiring during early brain development through its regulation of protein synthesis at the synapse. Advances in translational research at each end of this spectrum will uncover new candidate signaling pathways to exploit for therapeutic purposes. It is also important to consider that FXS is not solely a disorder of cortical pyramidal neurons and that both affected individuals and Fmr1 KO mice exhibit anatomical or physiologic dysfunction in other brain regions. Another important aspect of spine dysgenesis in FXS and in other types of mental impairment is that unlocking its mysteries in any one of these disorders will probably benefit research into all forms of autism and intellectual dysfunction.
Proposed Experiments and Predictions
Footnotes
Acknowledgements
I am grateful to Drs. Alberto Cruz-Martin, J. Tiago Gonçalves, Gary Bassell, Anis Contractor, Peter Kind, and Anthony Holtmaat for their comments and suggestions on this review.
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by FRAXA, the Dana Foundation, and the NICHD/NIH (grant R01HD54453).