
Abstract
Why do memory abilities vary so greatly across individuals and cognitive domains? Although memory functions are highly heritable, what exactly is being genetically transmitted? Here we review evidence for the contribution of both common and partially independent inheritance of distinct aspects of memory function. We begin by discussing the assessment of long-term memory and its underlying neural and molecular basis. We then consider evidence for both specialist and generalist genes underlying individual variability in memory, indicating that carving memory into distinct subcomponents may yield important information regarding its genetic architecture. And finally we review evidence from both complex and single-gene disorders, which provide insight into the molecular mechanisms underlying the genetic basis of human memory function.
Human memory is a genetically complex trait that likely involves interactions between genes and experience-dependent (environmental) factors. Throughout human history, survival has depended on accurate representations of remembered knowledge—for the food-gathering human, remembering, for example, spatial locations of food sources. Thus, from an evolutionary standpoint, it is clearly advantageous for memory to be a highly heritable trait. Accordingly, the anatomy and functional role of the critical neural circuitry underlying declarative memory—namely, the hippocampus and adjacent entorhinal and parahippocampal cortices—are largely conserved across species, despite considerable diversity in other brain regions (Manns and Eichenbaum 2006). Furthermore, although the type of information processed by cortical areas may vary among species, intrinsic computations of the hippocampal system may not (Manns and Eichenbaum 2006). Yet, despite evidence for evolutionary conservation and heritability in humans, the specific genes and genetic networks that influence memory are largely unknown.
Many neurological and psychiatric illnesses associated with memory dysfunction (e.g., Alzheimer disease [AD] and other forms of dementia, schizophrenia, and multiple sclerosis) are at least partially under genetic control. As such, a more complete understanding of the genetic architecture and neural underpinnings of long-term memory could provide clues about the biological pathways that influence these disorders. To the extent that declarative memory is sensitive to the function of genes that also predispose to these illnesses, quantitative indices of memory function (i.e., memory “endophenotypes”) could be used, either independently or in conjunction with clinical diagnostic information, to identify genes that confer disease risk (Gottesman and Gould 2003). This strategy has been successfully employed in the investigation of nonpsychiatric complex diseases, such as asthma and heart disease, which have used quantitative phenotypes (total serum immunoglobulin E [IgE] levels and lipid variation, respectively), to map quantitative trait loci (QTL) associated with disease risk (Waterworth and others 2010; Zhang and others 2003). Thus, the use of a quantitative allied phenotype or endophenotype—such as performance on a memory task—holds promise for gene identification for brain-related illnesses.
In this review, we consider evidence for the heritability of declarative memory and focus on empirical evidence for both common and partially independent inheritance of distinct aspects of memory function. Next we highlight some genetic studies that have used declarative memory as an endophenotype, along with findings regarding memory deficits in disease states. Collectively, these studies suggest that genetic variation—in the form of both common variants and rare mutations—may independently affect similar components of hippocampal circuitry, resulting in partially overlapping deficits that emerge from unique genetic roots.
Cognitive Components of Memory
Declarative, long-term memory (LTM) involves memories that can last as little as a few days or as long as decades. It differs structurally and functionally from working or short-term memory, which stores items for time frames on the order of seconds. Short-term memory involves a temporary potentiation of neural connections that can become LTM via rehearsal and meaningful association. LTM is typically divided into two categories: declarative (or explicit) memory and implicit (or procedural) memory (see Fig. 1). Declarative memory refers to memories that can be consciously recalled, such as factual knowledge and events. Declarative memory has two further subdivisions, episodic and semantic memory (Tulving 1972). Episodic memory involves memory for specific events in time and enables us to remember past experiences. Semantic or conceptual memory refers to general knowledge of facts, word meanings, objects, and people without connection to a particular event. Although memory for episodic events tends to be specific to an individual, semantic knowledge is largely shared across people within a given culture.
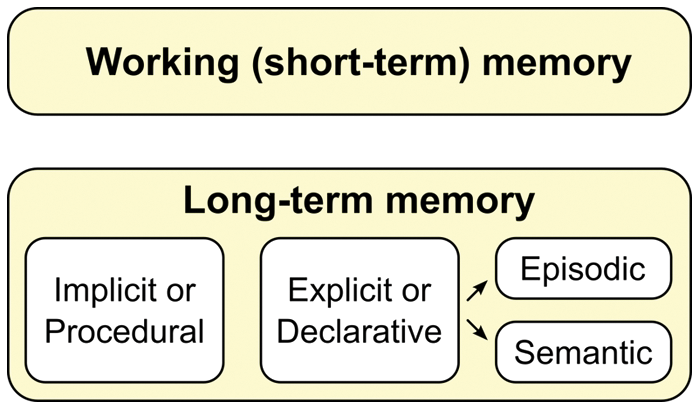
Long-term memory may be subdivided into categories, and it differs structurally and functionally from working and short-term memory.
Implicit or procedural memory (also known as nondeclarative memory) refers to unconscious memories for skills involving the use of objects or movements of the body, such as how to ride a bicycle. This type of memory involves neural circuitry that is dissociable from that involved in declarative memory, as it is associated with the basal ganglia rather than the temporal lobes (Gabrieli 1998). There is relatively little evidence regarding heritability of procedural memory; although the learning of motor skills is highly heritable (Fox and others 1996; Francks and others 2003), heritability of other forms of skills and habits has not been previously reported. These subtypes of memory can be differentially affected across disorders; for example, in AD, procedural memory abilities are relatively spared because these abilities are less dependent on prefrontal and hippocampal brain regions, which are most compromised with disease progression (Poldrack and Gabrieli 1997).
Assessment of Memory
Most behavioral probes of LTM involve the assessment of declarative rather than procedural memory. As with all cognitive domains, the memory literature represents a combination of standardized neuropsychological tests often employed as a part of clinical assessments and experimental tasks designed to address theoretical questions about memory function. Standardized neuropsychological tasks have the benefit of being comparable across study groups and populations but often test more general constructs. Experimental tasks have the benefit of being able to probe more refined questions about components of memory function but are not easily comparable across sites or studies (Barch and Carter 2008). Neuropsychological tests of declarative memory generally involve a delay between the encoding and retrieval (or recollection from memory stores) of information. In the verbal domain, list-learning tasks involving both an immediate and delayed condition are commonly used. Tasks of visual memory typically involve presentation of drawings or designs, which the subject must draw immediately and after a brief (approximately 20-minute) delay. To disentangle difficulties with encoding versus retrieval, memory tests typically include both a free recall (i.e., freely retrieving information from memory) and a recognition component (i.e., responding yes or no when presented with a particular item). In general, recognition is easier (less effortful) than free recall.
Neural Underpinnings of Memory
A crucial step in outlining the role of genetic influences on LTM is to understand the underlying functional neuroanatomy. Neural systems supporting declarative memory involve a set of interconnected neural networks linking neocortex, parahippocampal regions (including both perirhinal cortex and the more posterior parahippocampal cortex), and the hippocampus (see Fig. 2). Lesions to this region result in global amnesia characterized by an inability to form new memories and a temporally graded loss of previously acquired memories. The predominant contemporary view of this system highlights the role of more ventral regions of neocortex and closely connected perirhinal cortical regions that they project to, in the immediate representation of stimuli and maintenance of those representations over brief delays (Eichenbaum 2000; Wang and Morris 2010). More dorsal regions of neocortex and closely connected parahippocampal cortex participate in the representation and maintenance of stimulus context. Both of these constituents of the greater parahippocampal region cooperate to help representation persist, buffer against interference, and provide a venue for an initial phase of feature binding across information modalities and brief time intervals. The hippocampus in turn provides an additional degree of association building, linking representations across longer spans of time, binding items into the context of a particular learning episode, and allowing generalization (and inference) between related learning episodes (Eichenbaum 2000). Critically, once these assemblies of neocortical representations and their relationships are encoded, reactivation of any part of the assembly triggers activation of the entire, bound set of representations, allowing for retrieval of complex memories based on only partial cues (Wheeler and Buckner 2004). The role of the neocortex is not restricted to sensory regions subserving stimulus feature representation, however. Heteromodal regions such as prefrontal and parietal cortices also contribute to the conscious, effortful organization of information to be encoded, as well as to conscious recollection of learned information (while suppressing irrelevant information) and judgments based on the retrieved information.
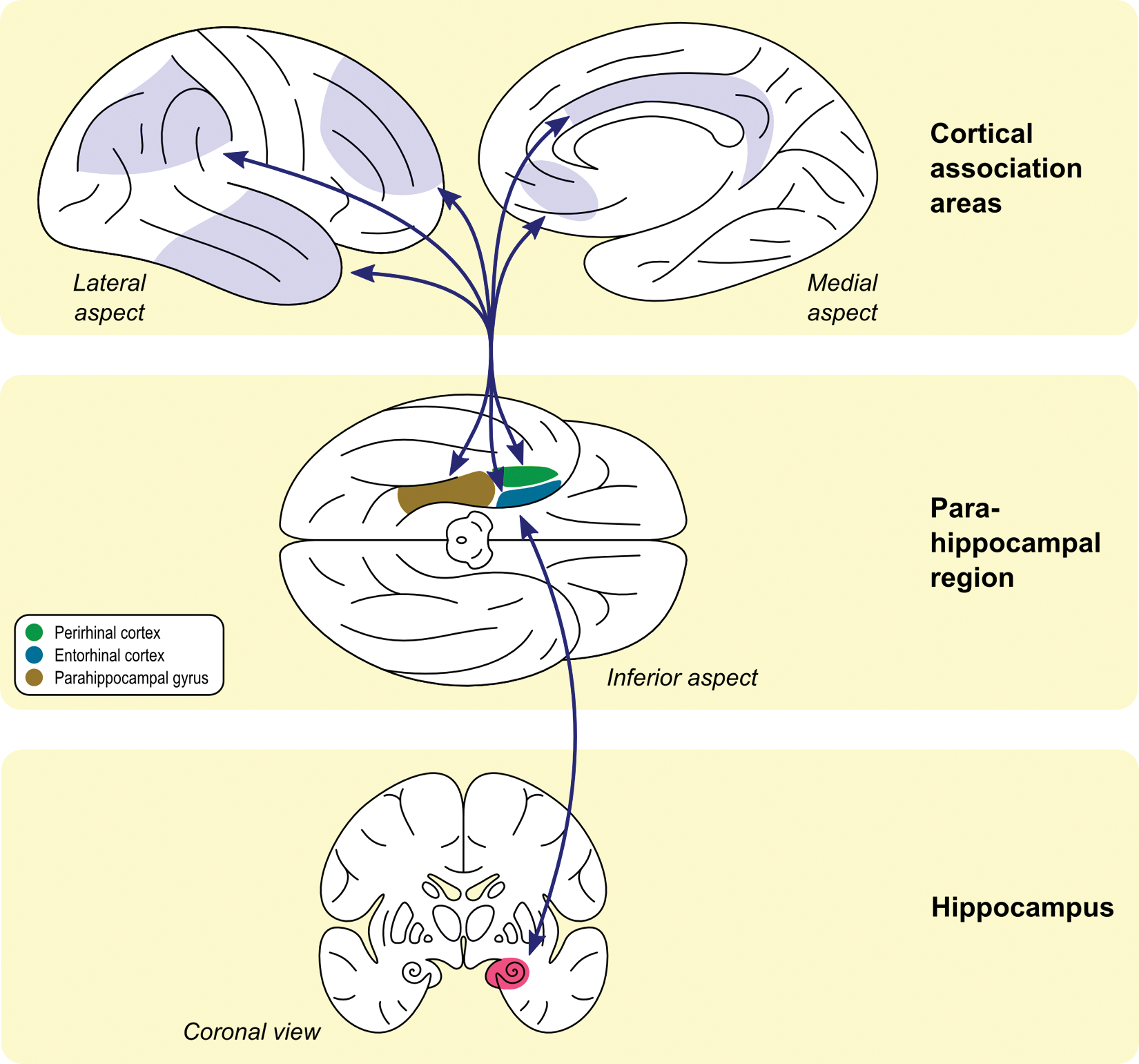
Information from multiple cortical association areas converges on areas that surround the hippocampus—namely, entorhinal, perirhinal, and parahippocampal regions. These regions are interconnected and project to the hippocampus itself. Efferents from the hippocampus reach the surrounding areas and then project back to the same cortical regions from where the inputs were originated (adapted from Eichenbaum 2000).
Positioned at the highest level of this associative hierarchy, the hippocampus displays a heterogeneous organization that facilitates its role in declarative memory. Information generally flows from perirhinal and parahippocampal cortices to the entorhinal cortex, which projects to the dentate gyrus, thus entering the hippocampus proper (see Fig. 3). The dentate, in turn, projects excitatory links to area CA3, which excites area CA1. CA1 then sends inputs to deeper, outgoing lamina of the entorhinal cortex, via the subiculum (Amaral and Lavenex 2006). Both intrinsic and extrinsic inhibitory connections innervate all levels of this system. Wang and Morris (2010) have proposed that the projections into CA1 provide information establishing spatial context for stimuli to be remembered, while separate inputs into CA3, and then into CA1, index the stimuli themselves. Building of associations within CA1 is therefore fundamental for binding objects and context information into the complex representation of a learning episode.
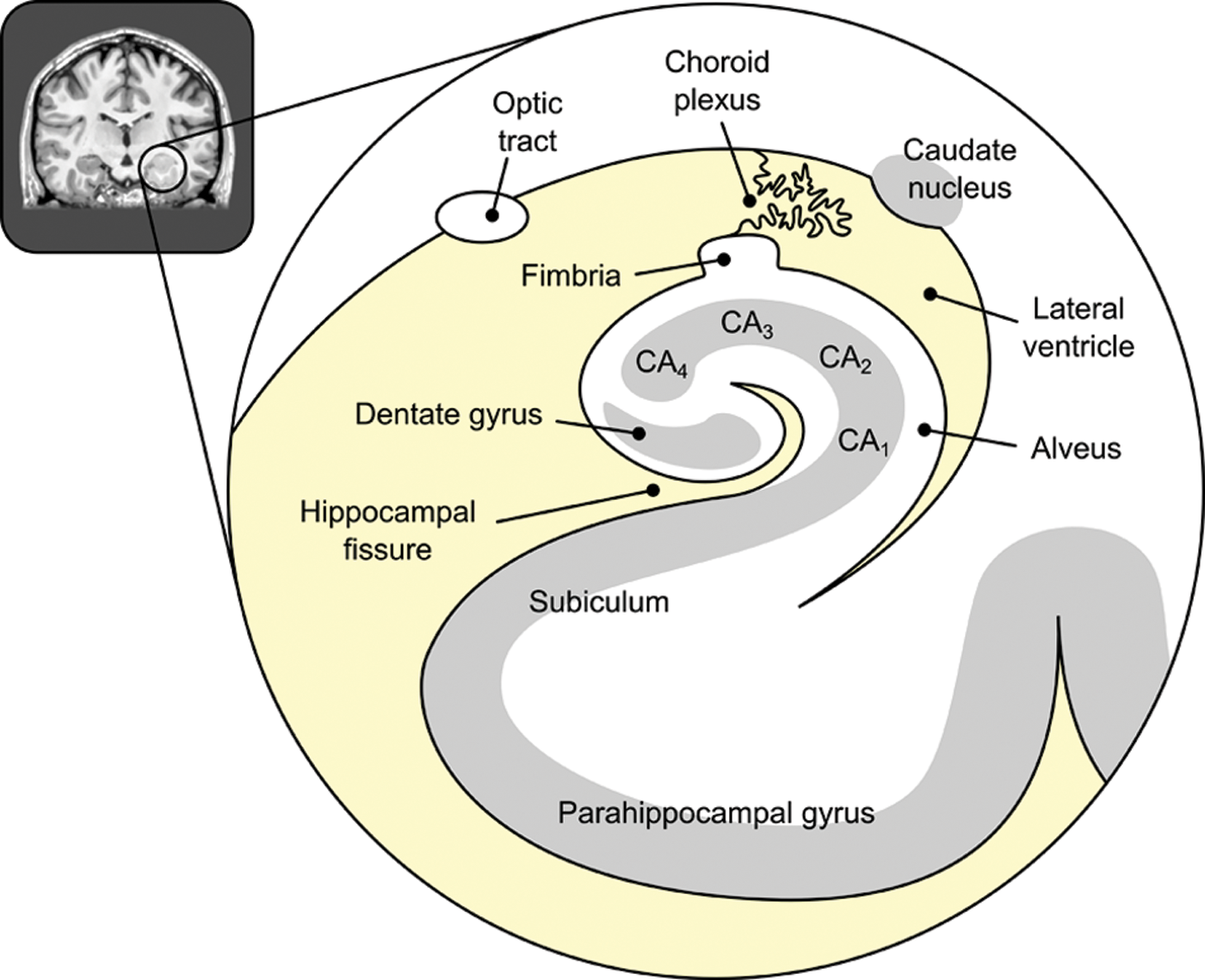
Schematic illustration of the main structures within and surrounding the hippocampus, as seen from a coronal slice through its anterior part.
List of Terms
In light of the highly associative nature of the process by which declarative memories are constructed and reconstructed, the creation, stabilization/consolidation, and reactivation of these associations are critical. Each of these phases, as well as each component of the circuitry they rely on, represents a point of vulnerability. Disruption of any of these components could result in a memory deficit, and a fine-tuned cognitive dissection of the deficit is needed to determine precisely which aspects of the process are impaired. As discussed below in relation to memory deficits associated with neuropsychiatric disease, genetic influences on any one of these processes could all result in a final common pathway of poor performance on a memory task, albeit through distinct mechanisms.
Molecular Basis of Memory
At the neuronal level, modification of the strength and efficiency of synaptic connections by synchronous activity in pre- and postsynaptic neurons—what Hebb famously postulated to be the cellular basis of memory—provides a model of the type of experience-dependent neural plasticity required of a physiological substrate of LTM. Although this modification can occur through a number of mechanisms (Malenka and Bear 2004), the most intensively studied example is long-term potentiation (LTP), or persistent enhancement of postsynaptic signaling triggered by a specific activity pattern during initial learning (Bliss and Collingridge 1993).
Perhaps the most widely studied example of LTP involves enhancement of CA1 pyramidal neuron activity after stimulation of presynaptic CA3 neurons—a phenomenon mediated by glutamatergic N-methyl-
Despite parallels between certain properties of molecular mechanisms of LTP and behavioral observations of fundamental aspects of LTM (e.g., fast initial encoding/modification, persistence over a year or more, stability amid interference), proving definitively that synaptic plasticity is necessary and sufficient for LTM functioning has been difficult (Neves and others 2008). Nevertheless, progress is ongoing as investigators strive to understand the molecular and cellular underpinnings of remembered experience.
Heritability of Memory
Twin and Family Studies
Heritability provides an index of the extent of genetic control over a trait. Heritability estimates (h2) range from zero, indicating no genetic contribution to trait variance, to 1, suggesting that the trait is completely under genetic control. Heritability is typically assessed through twin, family, and pedigree studies, in which the relationship between genetic proximity and similarity in cognitive performance indexes the degree to which a trait is associated with genetic or environmental factors. These designs have been frequently employed to assess the heritability of general cognitive abilities (Plomin and Kosslyn 2001). However, it is important to note that heritability estimates reflect the magnitude of the overall genetic effect on a trait but not the total number of genes that might be involved or their relative contributions (Almasy 2003). Thus, although higher heritability suggests that a trait is more strongly influenced by genetic factors, it does not elucidate the genetic architecture of the trait (e.g., whether it is influenced by many genes of small effect or a single gene of large effect).
Recent studies of genetic influences on cognitive abilities have highlighted the role of g, or general intelligence, based on findings of genetic correlations across different cognitive domains that are present even in cognitive disorders. The heritabilities of individual cognitive traits, including components of LTM, have been investigated, although not to the same extent as general intellectual function (IQ). There is evidence that episodic memory performance in young to middle-aged adults is moderately heritable, with estimates falling around 50% (Finkel and McGue 1993; Swan and others 1999), suggesting that half of all between-subject variation in memory performance is due to genetic factors. However, various component processes of memory may have different genetic determinants. For instance, a recent study of middle-aged twins showed evidence for shared genetic influences on learning ability and retrieval (h2: 0.36), as well as unique genetic influences on learning ability, likely reflecting unique genetic contributions to information acquisition (Panizzon and others 2011).
There is also evidence that heritability estimates for episodic memory are heavily task dependent. For instance, strategy use is genetically influenced; therefore, the degree to which a measure is amenable to strategy use may mediate the heritability of episodic memory performance (Nandagopal and others 2010). Factors such as processing speed constraints may also account for some of the genetic influence on episodic memory, particularly as related to the heritability of age-associated changes (Finkel and others 2009). In addition to the observed heritability of behavioral assays of memory, twin and family studies have shown the neuroanatomic structures associated with these cognitive functions to be heritable as well (generally with higher heritability estimates than behavioral measures). For instance, volumes of temporal regions have heritabilities in the range of 50% to 80% (see Tables 1 and 2).
Heritabilities of Memory and Nonmemory Cognitive Tasks
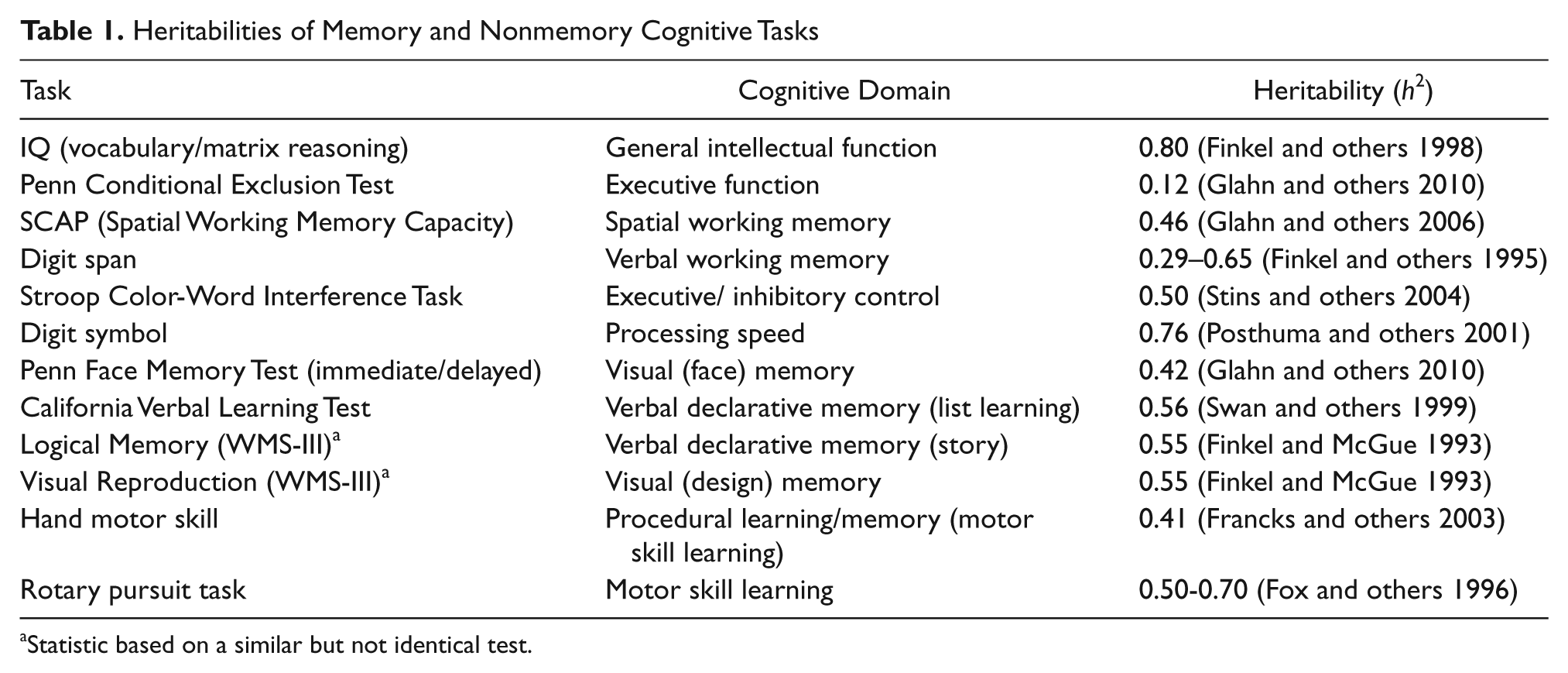
Statistic based on a similar but not identical test.
Heritabilities for Brain Structures Implicated in Memory
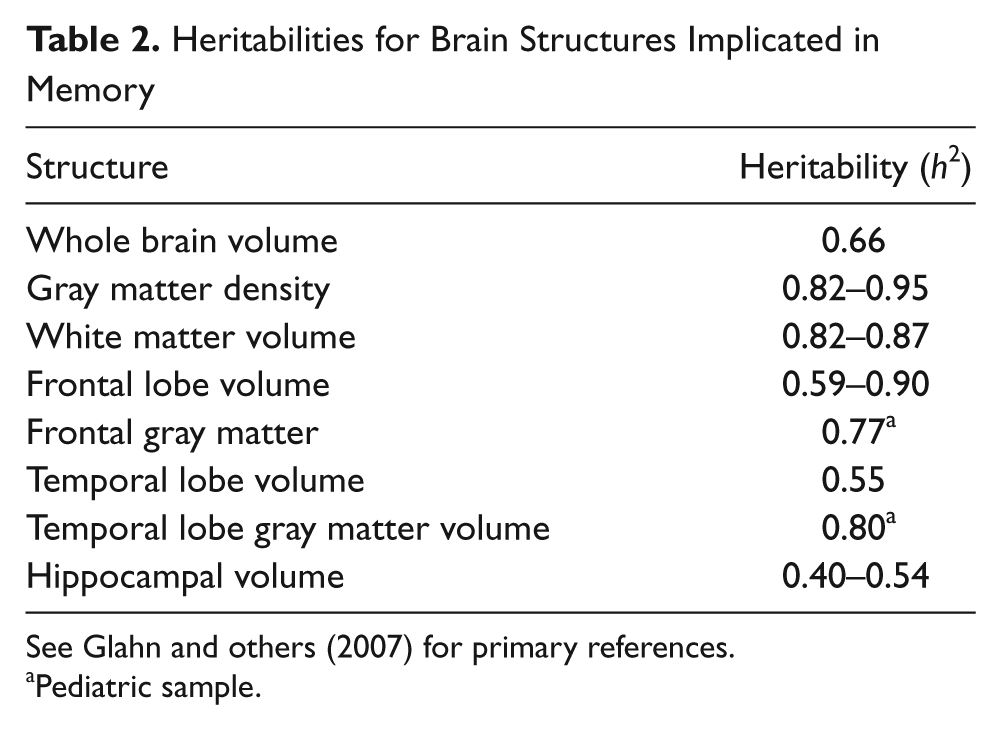
See Glahn and others (2007) for primary references.
Pediatric sample.
Specialist versus Generalist Genes: The Case for Faces
Although many genes are likely to contribute to common pathways that affect memory performance, there may also be specialist genes related to particular types of memories. Evidence for such specialist genes comes from two recent studies of face recognition or face memory abilities, suggesting that face memory is both substantially heritable (Wilmer and others 2010; Zhu and others 2010) and may be qualitatively distinct from other kinds of visual memory. In a large study of healthy adult twins, additive genetic factors accounted for 68% of the total variation in face recognition performance and 100% of the familial resemblance; task performance was only modestly correlated with other visual and memory abilities, suggesting that both face recognition ability itself and its genetic underpinnings are largely domain specific (Wilmer and others 2010). These findings nicely complement those of Zhu and colleagues (2010), who found significant heritability for standard face recognition but not for recognition of houses or inverted faces. Data from singleton adults also revealed independence of upright face memory from both general cognitive abilities (verbal paired associate memory and IQ) and from nonface visual recognition for abstract art. In addition, evidence from prosopagnosia studies indicates that severe face recognition deficits can run in families independent of IQ, sometimes with normal recognition for nonface objects (Lee and others 2010; also see McKone and Palermo 2010). The neural mechanisms underlying face recognition—involving the bilateral midfusiform gyrus—are well established in both humans and nonhuman primates (Kanwisher 2006). As such, cognitive neuroscience studies may guide genetic investigations of this socially advantageous trait.
Genes Contributing to Normal Variability in Memory Function
Behavior genetics studies are informative regarding the relatively large contribution of genetic factors to memory function but cannot inform us about the specific genes involved. In the past decade, increasingly high-density genotyping platforms have afforded an opportunity to examine the genetic basis of human memory on a genome-wide level. A genome-wide screen (Papassotiropoulos and others 2006) found that a locus encoding the KIBRA gene was significantly associated with performance on a verbal learning and memory test in a Swiss cohort of 351 healthy young adults. This finding was subsequently validated, using two slightly different verbal memory tasks, in a sample of outbred, cognitively normal adults from the United States. There were no allele-dependent differences in performance on control tasks of executive function, attention, or working memory, suggesting that the action of the KIBRA gene is specific to hippocampal-dependent memory. This finding was replicated in a second Swiss cohort, using a visual episodic memory task. The investigators further bolstered the association findings by fine-mapping the genomic region harboring KIBRA and the flanking genes, to ensure that the observed association was not due to linkage disequilibrium with surrounding genes. Next, they determined expression levels of KIBRA in the hippocampal formation and dentate gyrus in both human and murine brain tissue, finding that expression levels of the truncated KIBRA protein were higher in these structures than in other, non-memory-related brain structures. Finally, fMRI studies of an associative memory task were conducted in a subset of Swiss participants. Although there were no allele-dependent differences in encoding or in behavioral performance, during the retrieval phase, individuals without the T allele showed significantly increased neural activity compared with T allele carries in the medial temporal lobe and frontal cortex, suggesting that they require more activation in memory-related brain structures to achieve comparable performance.
The KIBRA protein is known to act as a binding partner for dendrin, a putative modulator of synaptic plasticity, and also to interact with multiple proteins involved in vesicular transport and neuronal plasticity (Schneider and others 2010). Although subsequent studies in other populations have not replicated this association (Need and others 2008), Papassotiropoulos and colleagues’ (2006) study is unique in its multilevel, mechanistic investigation of how the KIBRA gene may be involved in the neurobiology of memory. A recent study from the same group has investigated the role of this gene in AD, suggesting a modest role for this gene’s involvement in memory and AD risk (Corneveaux and others 2010).
Genes Contributing to Memory Deficits in Disease States
Disturbances of memory are a central feature of a number of psychiatric and neurological illnesses and often appear to be genetically mediated endophenotypes for the disorders (see Fig. 4). One line of evidence for genetic influence comes from family studies demonstrating the presence of deficits in unaffected relatives who carry some risk genes but do not suffer from the clinically manifest disorder. AD and schizophrenia represent two salient examples.
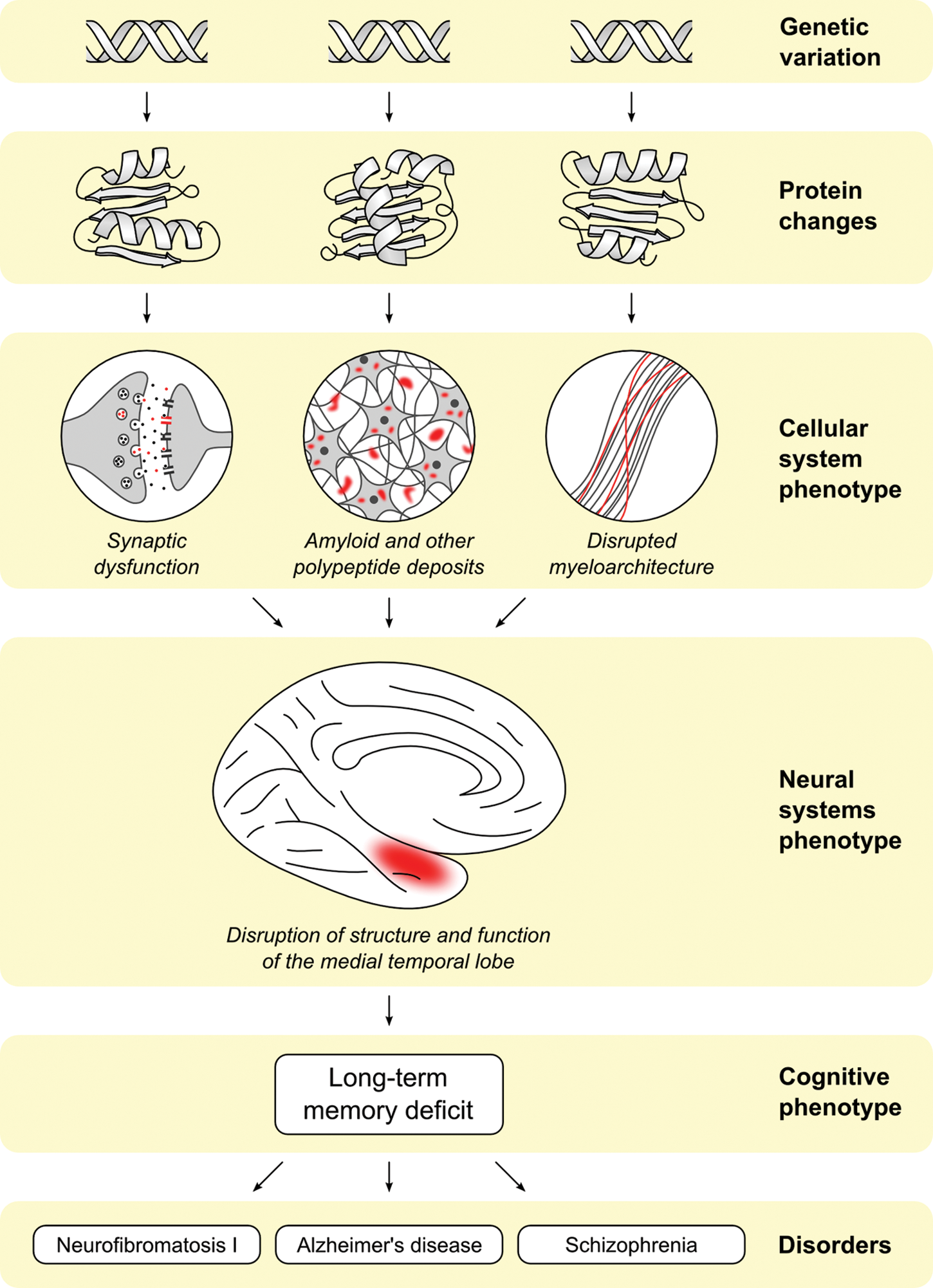
Long-term memory (LTM) deficits are a central feature of multiple disorders (both complex and Mendelian). LTM may serve as an endophenotype for these disorders, as the downstream expression of multiple underlying genetic, cellular, and neural systems abnormalities.
Alzheimer Disease
AD is a neurodegenerative disorder that affects approximately 3% of the population. Heritability of memory phenotypes in the context of cognitive disorders is complex, as both genetic and disease-associated factors are involved. As a result, there may be less familial resemblance in the memory phenotype (i.e., low sibling correlations) if a major gene with nonshared environmental influences underlies the phenotype. For instance, heritability of episodic memory performance among unaffected family members of AD patients was found to be 0.62, slightly above the range typically reported in younger healthy individuals (Wilson and others 2011). Furthermore, unaffected siblings show memory deficits relative to healthy age-matched controls, particularly when the ill sibling has an early onset form of AD (La Rue and others 1992). However, asymptomatic adult children of AD probands showed little evidence of similarity (i.e., low intraclass sibling correlations with heritabilities close to zero) on memory tasks sensitive to Alzheimer symptomatology, as compared to other, nonmemory tasks that showed heritabilities ranging between 0.24 and 0.52 (Smalley and others 1992). However, a subgroup of siblings had markedly lower memory performance, with that subgroup putatively representing a prodromal AD group. So, the degree to which siblings resemble their affected relatives may depend not just on overall genetic proximity but on the mode of genetic inheritance and which particular genes are shared. In support of this, there is evidence that AD is genetically heterogeneous, involving not just additive risk based on genes of small effect but a few genes of large effect, such as apolipoprotein E (APOE), which has been strongly and unequivocally associated with risk for AD, accounting for 40% to 60% of the genetic susceptibility to the disease (Lunetta and others 2007), as discussed below.
In addition to behavioral studies, neuroanatomic phenotypes have also been examined in AD patients and their clinically unaffected siblings. These MRI phenotypes—specifically, cerebral and medial temporal atrophy, white matter hyperintensities, and indices of cerebrovascular disease—are heritable and may be useful endophenotypes for genetic studies. Heritability of these brain phenotypes remained significant after covarying for APOE genotype, implying that a substantial proportion of the additive genetic variance is explained by other genes (Lunetta and others 2007). In addition, healthy adults with a maternal history of AD show brain metabolic changes in temporal, parietal, and frontal regions that resemble those observed in the prodromal stages of the disorder (Mosconi and others 2007).
The APOE gene on chromosome 19 likely represents the most well-validated common genetic variant identified to date that is relevant to memory function. There are three possible allelic variations of APOE (2, 3, and 4), with 3 being the most frequent. The APOE-4 allele has been strongly associated with risk for AD, and the risk conferred is gene dose dependent, with those individuals carrying two APOE-4 alleles having the highest risk for developing the disease (Tsai and others 1994). In addition to its role in AD, the APOE-4 genotype is associated with a number of memory-related phenotypes such as smaller hippocampal volume, lower functional activation during memory tasks, and thinning in medial temporal regions (Donix and others 2010). Other studies have also reported that episodic memory declines in APOE-4 carriers prior to symptomatic presentation of mild cognitive impairment (Caselli and others 2004).
Mechanistically, apolipoprotein E is involved in lipid metabolism. The APOE-4 genotype results in lower levels of APOE and consequently higher concentrations of circulating cholesterol (Takeda and others 2010). It is also associated with gene dose-dependent decreases in dendritic spine density in postmortem brains (Ji and others 2003). Relevant to AD neuropathology, the APOE-4 protein may be associated with tau and amyloid production, although not necessarily neurofibrillary tangles except in older subjects (Takeda and others 2010). In patients with AD, APOE-4 genotype is associated with a worsened course of the disorder, as well as more profound neural changes such as increased hippocampal atrophy, as shown in Figure 5 (Pievani and others 2010).
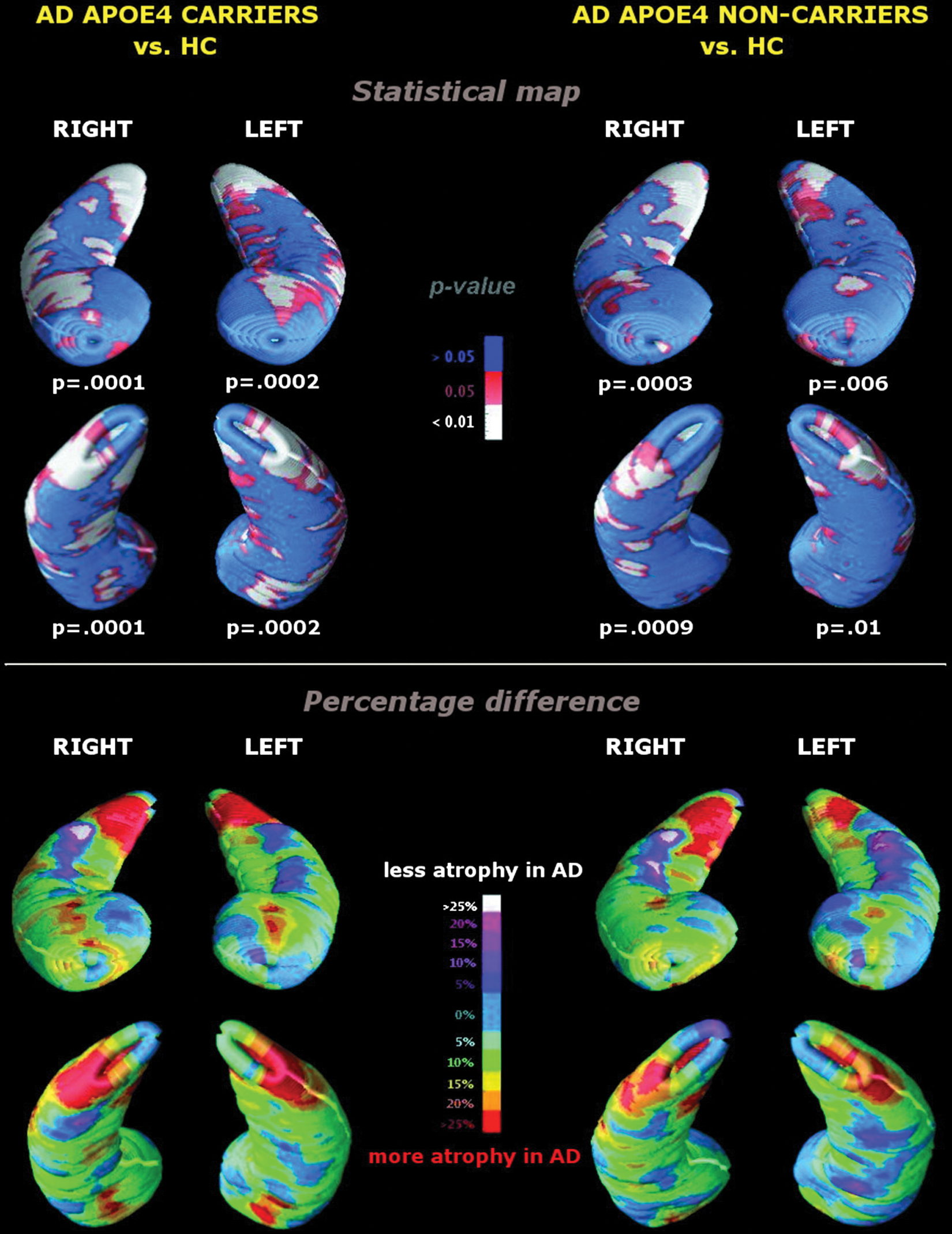
Patterns of hippocampal atrophy in Alzheimer disease (AD) patients with (left panel) and without the APOE-4 allele (right panel) compared with demographically matched healthy controls. Top: statistical maps. White regions correspond to an uncorrected threshold of P < .05. Comparisons were significant after correction for multiple testing by permutation testing, both in the ventral and dorsal hippocampal portions, bilaterally. Bottom: percentage hippocampal differences. Values are color-coded to express the percentage difference in radial size between AD patients and healthy controls. Values greater than 15% (yellow to red regions) denote statistically significant atrophic areas, and red regions correspond to areas of severe hippocampal atrophy (differences greater than 25%). From Pievani and others (2010).
Schizophrenia
As a broad phenotype, LTM deficits also present in schizophrenia and predict much of the disorder’s characteristic functional impairment (Green MF and others 2000). However, more fine-grained analysis demonstrates important differences in the cognitive profiles of schizophrenia versus AD. In particular, the severity of the mnemonic impairment in AD tends to be greater than in schizophrenia, involving impairment in both the encoding of novel information and the retrieval of well-learned information (i.e., “forgetting”). Schizophrenia patients, in contrast, tend to show slowed, inefficient learning of novel information but can generally retrieve that information once it has been consolidated (Cirillo and Seidman 2003). In addition, although AD tends to emerge later in life and show a degenerative course, schizophrenia is a distinctly neurodevelopmental disorder, with cognitive impairments observable long before the emergence of frank psychotic symptoms (Reichenberg and others 2010), reflecting latent illness vulnerability.
Declarative memory deficits are also present in high-risk adolescents (Brewer and others 2005) and nonpsychotic relatives of schizophrenia patients (Faraone and others 1999), further suggesting that this cognitive phenotype derives at least in part from an inherited genotype. However, because these memory deficits are more pronounced in patients compared with their own healthy monozygotic co-twins, nongenetic, disease-related factors must also be involved.
As in AD, computational brain-mapping approaches have revealed underlying neural correlates of schizophrenia patients’ verbal learning and memory deficits, demonstrating both genetic liability and disease-specific effects on hippocampal volume. Studies comparing monozygotic (MZ) and dizygotic (DZ) twins have also allowed the distinction between unique environmental and unique genetic effects on endophenotypes of interest. Specifically, in a population sample of Finnish twins concordant and discordant for schizophrenia, hippocampal volume varied in a dose-dependent fashion with genetic loading for schizophrenia (see Fig. 6). However, intraclass correlations (ICCs) for hippocampal volumes among healthy MZ pairs were larger than those among healthy DZ pairs, but the ICCs for hippocampal volumes among MZ and DZ pairs discordant for schizophrenia were equivalent. Together, these findings indicate that although hippocampal volume in healthy subjects is under substantial genetic control, hippocampal volume in schizophrenia patients and their relatives appears to be more strongly influenced by unique and shared environmental factors (van Erp and others 2004). Furthermore, intrapair differences between patients and unaffected co-twins in hippocampal volume and declarative memory performance were highly positively correlated, indicating that these neural abnormalities are strongly tied to the behavioral phenotype (van Erp and others 2008). Functional neuroimaging of declarative memory performance among nonpsychotic, non-twin relatives of schizophrenia patients offers additional evidence suggestive of abnormal prefrontal and temporal lobe activity associated with memory recollection, presumably reflecting genetic vulnerability (MacDonald and others 2009).
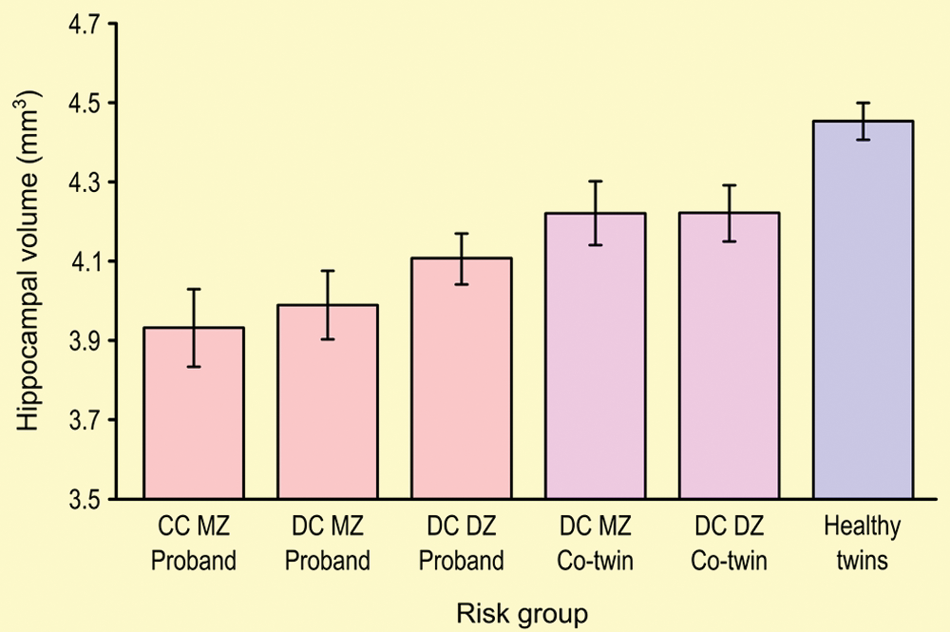
Mean hippocampal volumes and standard errors in subjects with a diagnosis of schizophrenia (probands), compared to their monozygotic (MZ) or dizygotic (DZ) twins, concordant (CC) or not (DC) with the diagnosis, and compared with a group of healthy control twin pairs (MZ and DZ). Hippocampal volumes are reduced according to the putative genetic loading for the disorder. Adapted from van Erp and others (2004).
Neuropathology studies indicate lower neuronal size in schizophrenia patients, possibly fewer neurons of specific types, and lower levels of a range of presynaptic proteins, particularly in the terminal fields of projections from the entorhinal cortex (Sawada and others 2005). In addition, recently evidence suggests adult neurogenesis may be reduced in schizophrenia, which may contribute to impaired cortical-to-hippocampal connectivity (Reif and others 2006).
Although much work remains in identifying the genetic architecture of the mnemonic impairment in schizophrenia, there are some promising leads associated with the disorder’s distinctly developmental course. For example, the neuregulin (NRG1) gene has been linked independently to schizophrenia vulnerability (Stefansson and others 2002) and to the regulation of LTP in the hippocampus (Kwon and others 2005). Possibly, decreased NRG1 signaling may perturb the activity-dependent maturation of AMPA receptors, in turn degrading the development of hippocampal NMDA receptors in a manner that would mimic the developmental course of schizophrenia (Li B and others 2007). Imaging studies have linked the NRG1 genotype to modulation of neural activity during episodic memory encoding and retrieval in healthy individuals (Krug and others 2010). However, examination of more than single-nucleotide polymorphism (SNPs) within particular candidate genes is needed to advance our understanding of the mechanisms by which neuregulin may affect memory function.
Neurofibromatosis Type I
A complementary approach to the search for susceptibility genes for complex traits such as memory involves the study of specific chromosomal mutations associated with memory dysfunction. Importantly, this approach facilitates the use of translational models, as the genetic cause of the disorder is well characterized. Mouse models for individual candidate genes allow the investigation of the function of these genes and how they may affect specific cognitive and neural phenotypic features. As such, single-gene disorders have dramatically enhanced our understanding of the molecular mechanisms of memory and how these mechanisms are perturbed in the context of particular genetic mutations (Bearden and others 2008).
Neurofibromatosis type I (NF1), or von Recklinghausen disease, affects 1 in 4000 people worldwide, making it one of the most common single-gene disorders affecting learning and memory in humans. This disease is caused by mutations in the NF1 gene on chromosome 17q11.2, which encodes neurofibromin, a rat sarcoma viral oncogene homolog (Ras) GTP-ase activating protein (GAP) that is highly expressed in the brain and has a key role in modulating hippocampal inhibition during learning (Brannan and others 1994). There is substantial evidence implicating the Ras signaling pathway in synaptic plasticity and LTM formation. Studies of mice with a heterozygous-null germ-line Nf1 mutation (Nf1+/– mice) have shown that these animals have enhanced inhibitory transmission, which is likely mediated by enhanced release of GABA, the main inhibitory neurotransmitter in the central nervous system. This increased inhibition seems to directly cause deficits in spatial learning in the Morris water maze (Costa and others 2002), a hippocampal-dependent task, and deficits in LTP. Other hippocampal physiologies are unaffected by the Nf1+/– mutation, suggesting a relatively selective effect on LTP. The spatial learning deficits of Nf1+/– mice closely resemble those observed in human NF1 patients (Shilyansky and others 2010), suggesting that the mouse model is highly relevant to the human condition.
Costa and colleagues (2002) also found that the LTP deficits in these Nf1+/– mutant mice can be rescued by genetic and pharmacological manipulations that decrease Ras gene function, indicating that the learning deficits associated with NF1 are likely caused by excessive Ras gene activity. These models have important implications for the development of targeted treatments for memory dysfunction. For example, memory deficits associated with Nf1 mutations can be reversed in adult mutants with a brief treatment of farnesyl transferase inhibitors (Li W and others 2005). Insights into the mechanisms responsible for NF1 may result in the development of sustainable treatments for this disorder and other disorders involving learning and memory deficits caused by dysfunction in this signaling pathway.
Common versus Rare Genetic Mechanisms
Although neurofibromin and APOE genes work through different mechanisms, they do show convergence on a final common pathway of hippocampal disruption. In NF1, this takes the form of increased GABA-ergic inhibition and resulting decreases in hippocampal LTP. The effects of APOE-4 genotype in mice involve decreased dendritic spine density in primary hippocampal neurons (Dumanis and others 2009). The overlap of these neuronal changes may provide a mechanistic explanation for shared memory phenotypes across neuropsychiatric disorders. At the same time, subtle phenotypic differences may be useful for dissecting the underpinnings of these complex disorders.
Methodologic Considerations: Polygenicity, Genetic Pleiotropy, and Phenotype Definitions
How does variation in a gene affect the expression of that gene and the functioning of the gene product? The genetic basis of LTM is clearly complex, likely involving similar phenotypes based on the combined actions of several (perhaps hundreds) of different genes together with environmental exposures. Each individual gene may have little effect on the disease phenotype (Bearden and others 2009). Gene-mapping techniques for disorders with simple inheritance patterns (i.e., Mendelian disorders) depend on the fact that within a family, individuals displaying the same affected phenotype could be assumed to possess the same genetic mutation. Clearly, different genetic analysis techniques are required for complex, multiply determined traits. The main approaches for genetic investigation—linkage and association studies—can be differentiated in terms of their focus on either variants of large effect or variants of small effect. The feasibility of these different approaches has largely been based on the opportunities for assaying common or rare genetic variants in well-powered studies. In addition, although most genetic research has focused on single-nucleotide alterations, it is now clear that structural copy number variation occurs more frequently than was previously realized and that such variation may be important in generating disease phenotypes.
As a highly complex trait, there are likely many genes in which functional variation can affect aspects of memory (polygenicity) (see Fig. 7). Conversely, genes likely affect neural networks and multiple brain functions, not isolated brain regions (Green AE and others 2008). As such, a single gene can be involved in multiple cognitive (and perhaps noncognitive) processes (pleiotropy). Thus, a systems approach is necessary to understand pleiotropic effects on cognitive functions, including memory. For example, examining genetic correlations between multiple brain structures and multiple cognitive processes can provide a window into the shared genetic variance across neural circuits and cognitive domains, as well as unique genetic influences on particular, sometimes highly correlated, neurocognitive processes (e.g., Panizzon and others 2011).
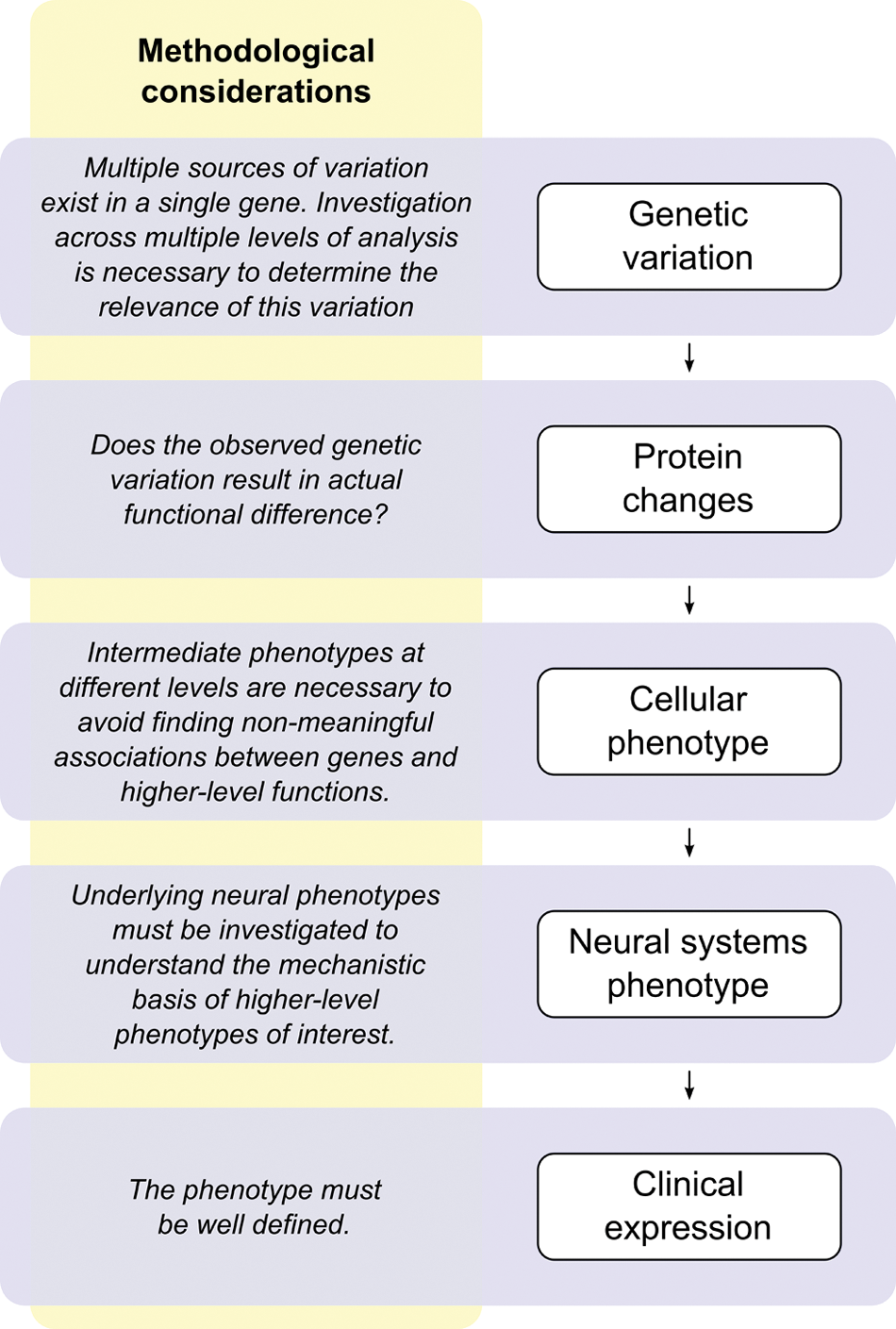
Complex traits or disorders, such as memory or Alzheimer disease, which influence and are influenced in multiple ways, can only comprehensively be understood by disentangling their mechanisms at each expression level. Successful research at any level of analysis must build on discoveries on all other levels.
Although molecular-genetic data are essential for understanding how a genotype connects to a disease phenotype or associated intermediate phenotype, attempts at gene identification will fail without well-defined phenotypes. Interpretation of measures and/or specific cognitive abilities showing low heritability is confounded by variation in the psychometric properties of cognitive tasks. Thus, rigorously defined psychological constructs are critical for moving the field forward. The same principles apply to traits derived from brain structure, where even seemingly objective choices of phenotypes can influence the results (Winkler and others 2010).
Future Directions
Genetic information has the potential to inform key questions related to the cognitive neuroscience of human memory. However, despite reasonably high heritability, thus far few replicable genetic associations have been identified for normal variation in memory function. Is the genetic structure of memory too complex to be tractable? Or, do we need to further refine the phenotypes we are using to interrogate more specific memory subprocesses? Further advancement in our understanding of the genetics of human memory will depend on further developing the theory and methods for defining memory phenotypes, at both the behavioral and neural levels, and characterizing the function of relevant genes and gene networks at the molecular level. Below are suggestions for specific innovations needed to move the field forward.
Translational studies in animal models provide a valuable means of interrogating the underlying biology of specific memory phenotypes. Memory deficits represent a scientifically tractable and physiologically plausible target for psychopharmacologic treatment. Rare structural mutations provide useful models, as the underlying genetic etiology is already known, and effects tend to be large. The cellular mechanisms modulated by particular genes of interest can be readily studied in rodent transgenic or mutant models. As evidenced by the development of a viable treatment for cognitive deficits in NF1, based on findings in a mouse model (Li W and others 2005), such models allow us to test pharmacologic agents that can reduce or attenuate memory deficits.
Improving methods for high-throughput cognitive phenotyping (for example, web-based assays of multiple memory functions) will allow studies to amass substantially larger samples than can be collected in the laboratory. One prominent example of such a study is the “Test My Brain” web-based testing environment, in which study participants consent and participate in cognitive testing entirely online (Wilmer and others 2010). This is a key innovation needed to accrue adequately powered samples to identify genes of small effect on memory function.
Finally, in the postgenomic era, one of the biggest challenges faced by interdisciplinary scientists is the lack of tools to manage the complexity of knowledge rapidly being amassed across disparate methods, models, and data types (Sabb and others 2008). Informatics resources can advance the collation of empirical knowledge that will help to bridge the currently wide gap between genome, cognitive constructs, and disease syndromes (Parker and others 2009). Such tools can advance our understanding of the genetic architecture of memory by helping researchers to identify previously unsuspected relationships across disciplinary boundaries, select specific phenotypic measures, and develop multilevel models that specify both within- and between-level associations (Fig. 8).
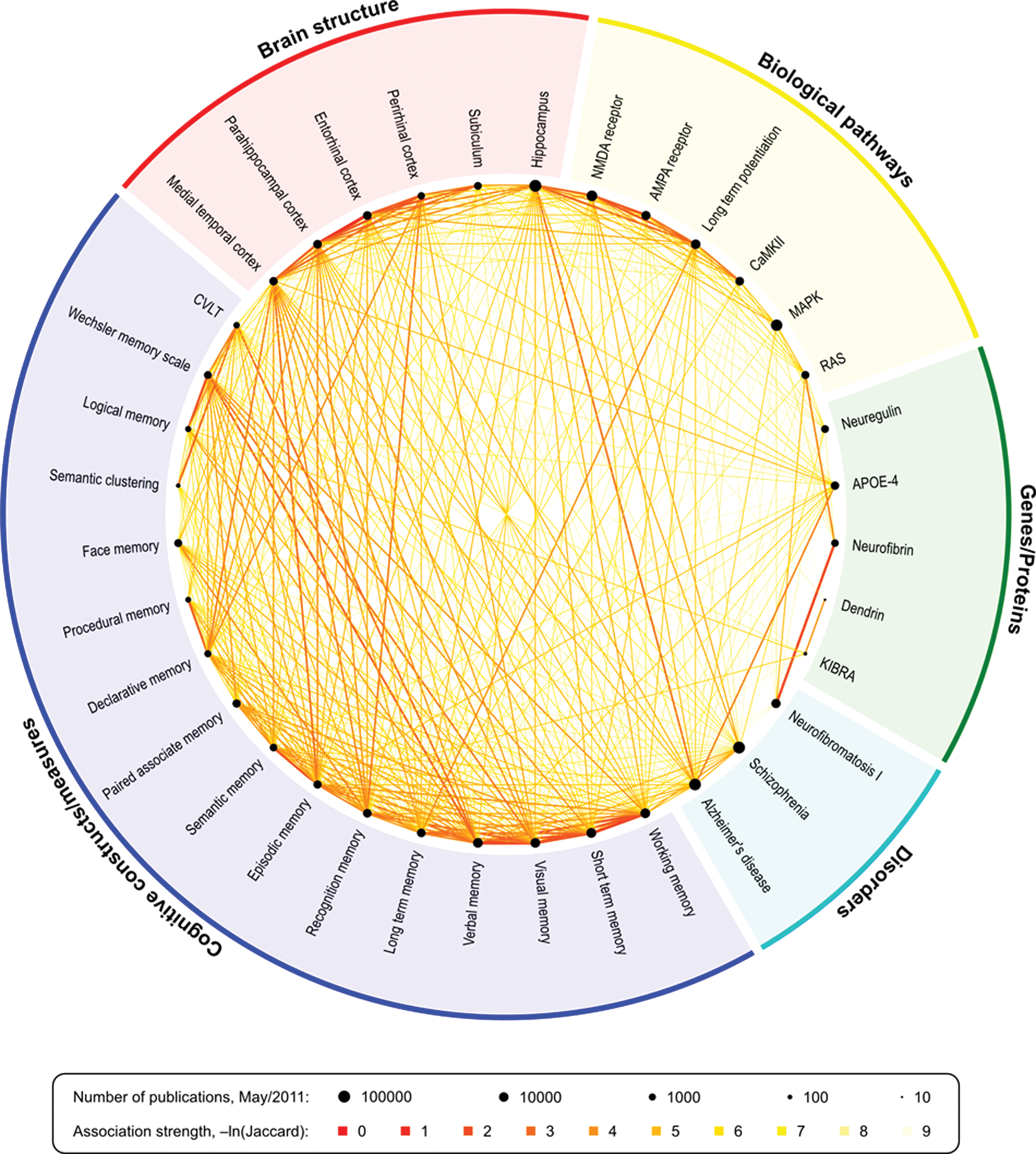
Relationships between keywords in published literature can expose interesting links between fields and highlight areas where important pieces may be missing, as well as evidence-emergent patterns of research. In this figure, each point around the circle represents the relative quantity of publications of a given keyword in relation to the others, in logarithmic scale, as retrieved from PubMed in May 2011. The links represent the strength of the association, scaled by the natural logarithm of the Jaccard coefficient. The smaller numbers indicate stronger associations. See also the Supplemental Material for an interactive depiction of these relationships and http://www.pubatlas.org/ for more literature mining and visualization tools. NMDA = N-methyl-
Conclusions
Given the central importance of learning and memory to adaptive behavior, there are likely multiple—possibly even hundreds—of genes of small to moderate effect that influence memory phenotypes in the general population. Many of these genes may contribute to disease susceptibility via their impact on brain systems mediating memory function; as such, these genes may not have been identified by previous studies using syndromic status (e.g., “schizophrenia”) as the phenotypic target. From a neural systems standpoint, it is plausible that cognition has both domain-general and domain-specific heritable contributions. Examining associations between genes and intermediate phenotypes will help to strengthen evidence for biological connections between genetic mechanisms and memory disorders; this mechanistic approach will also help to reduce spurious associations (Green AE and others 2008). As discussed here, investigation of memory phenotypes expressed across multiple syndromes and species, using an interdisciplinary, systems-level approach, will accelerate the discovery of new treatments for memory dysfunction.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
Financial Disclosure/Funding
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was partially supported by funds from the National Institute of Mental Health (grant numbers R01-MH080912-03, R01-MH078143-06, RL1-LM009833, RL1-DA024853, K23-MH74644, R34 MH08929).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.