
Abstract
In this article, we propose an X-linked hypothesis of brain disorders that postulates a neuronal origin of those neurodegenerative and psychiatric disorders with a greater male prevalence. The hypothesis is based on the accumulated genetics and genomic evidence linking X chromosome genes and transcripts to neuronal cells. The behavioral genetics literature has long pointed to the link between postsynaptic protein complexes coded on chromosome X and mental retardation. More recently, novel genomic evidence has emerged of X-linked mRNA overexpression of neuronal source in the human brain. We review the evidence for this hypothesis and its consistency with the distribution across genders of brain disorders of known aetiology. We then provide examples of the utilization of this hypothesis in the investigation of the pathophysiology of complex brain disorders in both the stratification of disease cohorts and the development of realistic preclinical models. We conclude by providing a general framework for testing its validity, which will be exploited in future studies, and provide future directions for research.
Background
Mendelian inheritance patterns are well documented in neurodegeneration; yet they are limited to a small number of disorders that are either strictly inherited, for example Huntington’s disease, or rare inherited forms of Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) (La Spada and Ranum 2010). Although neurodegenerative disorders are mostly sporadic, their clinical presentations and neuropathological findings resemble those of hereditary diseases. Hence, genetic linkage-analysis has been used to discover common pathophysiological mechanisms. Likewise, the majority of common psychiatric disorders show high genetic familiarity, with heritability estimated as high as 80% in schizophrenia (Sullivan and others 2003) and 40% in depression (Sullivan and others 2000).
However understanding the cellular mechanism by which genetic variation increases risk in neurodegenerative and mental disorders has long been elusive.
Classic positional candidate approaches to gene discovery employ linkage analysis to map a trait and then search for likely gene candidates in the regions that have been identified. Although this has been effective for monogenic traits, it has had limited success for complex phenotypes that are polygenic as well as influenced by environmental factors. One reason is that the function of most genes is still unknown. Another is that mapping resolution is usually severely limited (Georges 2007). Genome-wide association studies have identified thousands of genetic loci that contribute to common diseases in humans. However, these loci have typically modest value for predicting future disease occurrence and generally provide little mechanistic insight particularly as it may not be easy to understand how alleles interact with each other or with environmental factors. Studies using experimental models have shown that natural phenotypic variation usually results from a number of interacting alleles that produce context-dependent effects, hence the belief that common diseases will be similarly complex (Ayroles and others 2009; Huang and others 2012).
To compound further the difficulty of the investigation into their causative mechanisms, both neurodegenerative and psychiatric diseases are also characterized by a significant delay between the primary unknown pathophysiological event and the onset of clinical symptoms. The greatest risk factor for neurodegenerative diseases is aging, and these disorders are in great part characterized by a late onset. However, the study of the rare genetic cases demonstrates that the time interval between the start of disease and the appearance of overt clinical symptoms may be quite significant; adults at genetic risk for autosomal dominant AD demonstrate very early presymptomatic alterations of functional and structural magnetic resonance imaging as well as cerebrospinal fluid and plasma biomarkers (Reiman and others 2012). Similarly, psychiatric conditions generally emerge in late adolescence or early adulthood but their origin may be as early as prenatal (Watson and others 1999).
Here genetic evidence is reviewed to understand the aetiology of brain disorders, but instead of pointing to proteins, pathways, or neurobiological processes, it is used to indicate the neuron as the source of the pathology in disorders with a marked male prevalence. The disentanglement from the complex web of genetic and environmental interactions of a specific cell type as the culprit of a disease may be quite valuable; it may help the stratification of pathological cohorts, inform more realistic animal models, and guide the development of early biomarkers, and functional imaging biomarkers in particular.
In this paper, the focus is on the emerging link between the X chromosome and the brain and, more so, between the X chromosome and the neuron. We first review the supporting historical DNA data mostly contained within the behavioral genetics literature and then summarize the novel evidence emerging from genomic studies. We then point to the strong association between the disorders of known neuronal aetiology and the male gender, while a balanced gender prevalence or a marked female prevalence is generally associated to non-neuronal sources (glial, hormonal, immune, or other gender interacting environmental variables). Finally, we consider possible implications of the X-linked hypothesis in a more speculative section where we use differences in the gender prevalence of complex brain disorders to gain new insights into their origin.
The X Chromosome and the Brain
The X chromosome has unique properties stemming from the evolution of sex chromosomes in mammals. Both X and Y chromosomes have evolved from a pair of autosomes, but while the original elements have been conserved on the X, the Y chromosome has lost almost all traces of the ancestral autosome, including the genes that were once shared with the X. The hemizygosity for X chromosome genes exposes males to recessive phenotypes that account for the large number of diseases that have been associated with the X chromosome (Ross and others 2005).
The link between the X chromosome and the brain is not new, but it has been mostly noted in the psychology literature. Chromosome X contains about 4% of the human genome but hosts a disproportionate fraction of genes associated with mental disorders and mental retardation in particular (around 60 vs. the 10 expected) (Ropers and Hamel 2005). This cumulative evidence has led to the general theory, first proposed by Lehrke (1972), that major genes for intellectual function are located on the X chromosome. The implications of Lehrke’s proposal (e.g., inheritance of male intelligence via the mother, higher male vulnerability to mental disorders, higher prevalence of males in the highest and lowest percentiles of the IQ distribution given the overall mean IQ equality across genders), have been subject of interest and, unsurprisingly, controversy (Johnson and others 2009; Nisbett and others 2012; Turner 1996). However, although there is no argument that the X chromosome contains an enriched number of genes that, when mutated, cause intellectual deficiency, there is still no evidence that allelic variation along X contributes to normal range variation (Turkheimer and Halpern 2009) nor that X-linked genetic differences posits greater variance in cognitive ability in males (Giummo and Johnson 2012).
Recent advances of genomic technology have further supported the genetic data demonstrating a significantly higher density of X chromosome mRNA in the brain than in any other tissue (Nguyen and Disteche 2006). It is well known that while males inherit a single maternal copy of X, females inherit two copies from both parents, and hence in females one copy is randomly suppressed by a process called X-inactivation. In order to balance for the reduced amount of DNA of the single X chromosome compared to the two copies of the autosomes, a second transcriptional mechanism called dosage compensation has evolved to double the transcription of the active copy of X. Importantly, X dosage compensation seems to be tissue specific and exaggerated in the brain where the overall X:autosome mRNA ratio is >1.2 (Nguyen and Disteche 2006) (although the excess could be explained, at least in part, by the increased number of genes expressed in brain). It is important to note that approximately 15% of genes on the human X chromosome escape inactivation, as compared to 3% in the mouse, contributing to the emergence of sex differences (Berletch and others 2011). Finally, recent work in our lab has demonstrated that the excess X chromosome dosage in brain is exclusively of neuronal origin (Swingland and others 2012).
The genetic and more recent genomic evidence reviewed above supports the enrichment of neurons in the brain with DNA and mRNA from the X chromosome. This should result in a substantial increase of the risk for males, who have only one copy of the chromosome, of developing brain disorders of neuronal origin (Fig. 1). Conversely, diseases with a balanced gender ratio (or higher female incidence) should have non-neuronal aetiology as females are protected from X-linked illnesses because they have two copies of the chromosome. The next section reviews this hypothetical association in disorders of the CNS of known cellular etiology (see Figure 2).

The X-linked hypothesis of brain disorders is based on the recently observed enrichment of chromosome X derived genes of neuronal cells and postulates a neuronal origin of those neurodegenerative and psychiatric disorders with a greater male prevalence.
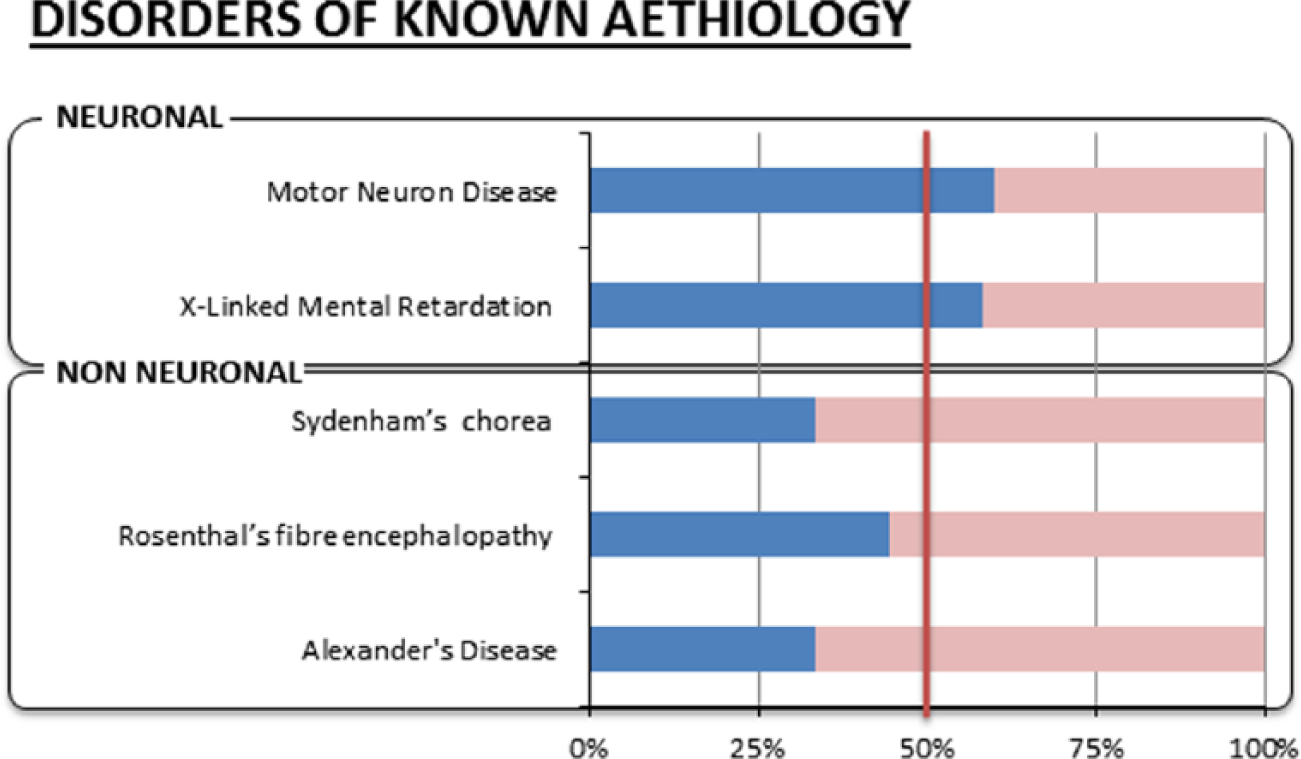
The evidence for the X-linked hypothesis consists in the greater male prevalence (in blue) of brain disorders with known neuronal aetiology, whereas disorders with known non-neuronal origin (glial, hormonal, immune, or other) have higher female prevalence (in pink).
Gender Bias and Brain Disorders of Known Aetiology
The greatest male-to-female incidence ratios for CNS disease are recorded for the X-linked mental disorders (1.4:1) (Ropers and Hamel 2005) where the most common form is Fragile X Syndrome, which has a male prevalence of 2:1 (Turner and others 1996). These disorders are linked to the neuronal multiprotein complexes and their components that are located on the X chromosome (Laumonnier and others 2007) and have been shown in animal models to be key to neuronal plasticity and cognitive processes (Laumonnier and others 2007).
Motor neuron diseases (MNDs) are also a set of disorders with significant difference in gender prevalence. MNDs include amyotropic lateral sclerosis (ALS), primary lateral sclerosis, progressive muscular atrophy, progressive bulbar palsy, and pseudobulbar palsy. These disorders are significantly most common in men (1.54:1 to 1.3:1) (Alonso and others 2009; McCombe and Henderson 2010) and have clear neuronal aetiology.
Rett syndrome represents an interesting contradiction as it is an X-linked disorder due to a mutation of the MECP2 gene but is present only in females (Amir and others 1999); however, the discrepancy is only apparent as the syndrome is symptomatic in females because it is milder, but it is fatal in males that usually do not survive to term or die within the first 2 years of life.
Conversely, two recognized primary astrocytic pathologies, Alexander’s disease and Rosenthal’s fiber encephalopathy (both characterized by cytoplasmic inclusions localized within astrocyte processes that are responsible for their metabolic damage and subsequent degeneration), show an increased prevalence in women compared to men (1:0.5 and 1:0.8, respectively) (Jacob and others 2003).
Equally, Sydenham’s or rheumatic chorea is a disorder that occurs mainly in childhood and twice as often in girls than in boys (Rabin and others 2014) and is considered an autoimmune neurological manifestation of acute rheumatic fever.
Gender Bias and Complex Brain Disorders
The data illustrated so far suggest that X-linked risk, the relative difference of incidence between men and women, points toward a neuronal pathophysiology if men are more at risk of the disorder or to a non-neuronal origin in the neutral or opposite case (e.g., when the incidence is the same or higher in women). This we define as the “X-linked hypothesis of brain disorders.”
In this section, we review the epidemiology of brain syndromes (see Figure 3), noting homogeneity or heterogeneity of gender prevalence within each diagnostic class, review known neuronal/non-neuronal mechanisms, and use, whenever possible, the X-linked hypothesis in a speculative fashion to fathom its potential to obtain novel insights into complex etiologies.
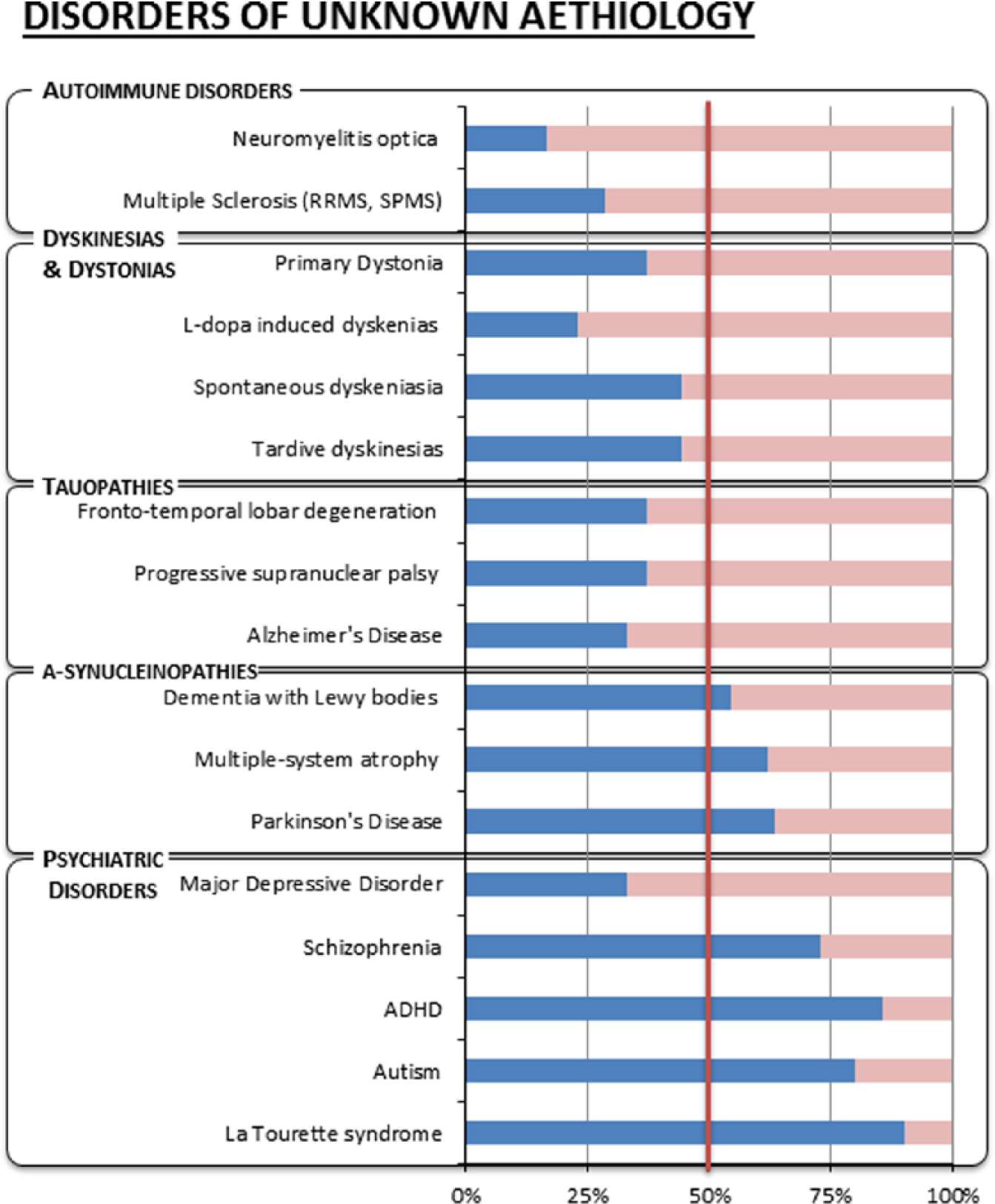
Male (in blue) and complementary female (in pink) prevalence of complex brain disorders of unknown aetiology.
Autoimmune Disorders
Multiple sclerosis (MS) is classified as an autoimmune disorder, and in keeping with the classification and the X-linked hypothesis, it is more common in the female population. However, this is strictly true only for the most common forms of MS such as relapsing-remitting MS (RRMS) and secondary progressive MS (SPMS). These forms of MS are associated with variable inflammatory response and affect women two to three times more than men (Alonso and others 2007). In a minority of patients instead, the disease assumes a noninflammatory progressive course and is termed primary progressive MS (PPMS). Recently, PPMS has been described as a separate disorder that is underlined by a primary neuronal cyto-degenerative aetiology (Stys and others 2012). The X-linked hypothesis supports this view given that the female prevalence noted in RRMS disappears and becomes a slight male preponderance in PPMS (Thompson and others 1997).
On the other hand, the gender ratio of diseases with supposed glial aetiology can also be interpreted in light of the X-linked hypothesis as in the case of neuromyelitis optica (NMO). NMO is an autoimmune disorder and a putative astrocytopathy characterized by inflammatory demyelinating lesions in spinal cord and optic nerve. NMO has an even stronger female predilection than does RRMS, with a female-to-male ratio that in some reports can be as high as 9:1 (Jacob and others 2013; Jarius and others 2012).
α-Synucleinopathies
PD is a common neurodegenerative disorder characterized by bradykinesia, resting tremor, muscular rigidity, and postural instability, as well as by a clinically significant response to treatment with levodopa. Epidemiological data suggest that male gender is one of the risk factors for the development of PD, with a lower incidence and higher age at onset in women (Haaxma and others 2007). Differences in the clinical manifestations and the course of PD have been observed between males and females, with the suggestion of a more benign phenotype in women. In women, the development of symptomatic PD may be delayed by higher physiological striatal dopamine levels, and they are also more often presented with tremor, which is associated with milder motor deterioration, concurrent non-dopaminergic pathology, and milder striatal degeneration (Haaxma and others 2007). Mutations in the α-synuclein gene (PARK1) have been found to result in autosomal dominant PD, and mutations in the parkin gene (PARK2) produce an autosomal recessive juvenile-onset form of disease. These genes are not X-linked, but genetic linkage studies for PD in samples without parkin mutations have consistently pointed to a strong linkage to the X chromosome (Morrison and others 2012; Pankratz and others 2002; Pankratz and others 2003; Scott and others 2001). Hence, in the context of PD, the X-linked hypothesis reinforces a causal link between a probable dopaminergic neuronal aetiology and X-linked genes.
It is interesting to note that all other α-synucleinopathies also display greater male prevalence. Multiple-system atrophy (MSA), a neurodegenerative disorder characterized by problems of movement, balance, and autonomic functions (regulation of blood pressure, bladder control, etc.), has been reported with higher male-to-female ratios, for example. 1.4:1 (Wenning and others 2004) and 1.9:1 (Swan and Dupont 1999).
Dementia with Lewy bodies (DLB), which is a type of dementia closely associated with PD with symptoms that include fluctuating cognition, visual hallucinations, sleep disorders, and PD related motor symptoms, is also more common in men than women (Rosenberg and others 2001). It is characterized anatomically by the presence of Lewy bodies, clumps of α-synuclein, and ubiquitin protein that develop in neurons (Dickson and others 1996).
Tauopathies
The application of the X-linked hypothesis to Alzheimer’s dementia would also indicate non-neuronal aetiology given the prevalence in females of this complex disorder (Bao and Swaab 2010). This is coherent with the E4 variant of apoliprotein E being the largest known genetic risk factor for late-onset sporadic AD in a variety of ethnic groups (Corder and others 1993), as the protein in the central nervous system is a product of astrocyte glia (Boyles and others 1985).
It is again of notice that gender balanced or increased female risk is shared by the other tauopathies. Progressive supranuclear palsy is another neurodegenerative disorder whose characteristics include gaze dysfunction accompanied by extrapyramidal symptoms and cognitive dysfunction. Reports state an equal prevalence between male and female (Nath and others 2001).
Corticobasal degeneration (CBD) is characterized by PD-related symptoms (bradykinesia and rigidity), limb dystonia, cognitive problems, apraxia, myoclonus, and dysphagia. The literature lacks proper epidemiological studies, but individual reports usually state predominant appearance of CBD in women. Notably, histological hallmarks of CBD are astrocytic abnormalities within the brain and excessive accumulation of the protein tau with high density of astrocytic plaques in the frontal lobe and premotor area of the cerebral cortex (Hattori and others 2003; Komori 1999)
Frontotemporal lobar degeneration (FTLD) is the second most common dementia after AD. It usually presents with significant changes in social and personal behavior and blunting of emotions. Both Japanese (Ikeda and others 2004) and European (Gilberti and others 2012) studies have reported that FTLD is more common in females. Here as well, the pattern of brain atrophy is underpinned by pyramidal cell loss and gliosis; however, at the early stages, neuronal apoptosis is rare, and it is the severity of astrocytosis and of astrocytic apoptosis that correlates with the disease stage and then becomes the major pathological feature as disease progresses (Broe and others 2004).
Dyskinesias and Dystonias
Dyskinesias and dystonias can be isolated or combined with other movement disorders, with or without a neurodegenerative substrate. Tardive and spontaneous are the common forms of dyskinesias. A meta-analysis of 76 studies has shown that the prevalence of tardive dyskinesias is significantly higher in women than in men (Yassa and Jeste 1992). This appears to be also the case with spontaneous dyskinesias (Merrill and others 2013).
L-dopa-induced dyskinesias (LIDs) instead may result from the combined effect of progressive neurodegeneration and consequent loss of buffering of exogenous L-dopa and usually appear in advanced PD following years of treatment with L-dopa.
Women also appear to have significantly greater prevalence of LIDs compared to men (Lyons and others 1998; Zappia and others 2005). Interestingly, it appears that men exhibit more significant classical PD symptoms (bradykinesia and rigidity), but women more LIDs. It is possible that genetic/genomic differences across gender may affect the signaling of D1 and D2 receptor mediated striatal pathways and, thus, regulate the differential response to L-dopa.
Dystonias are movement disorders in which sustained muscle contractions cause twisting and repetitive movements or abnormal postures. Dystonias can have both familial and environmental origins and are generally more common in women apart from writer’s cramp (Epidemiological Study of Dystonia in Europe Collaborative Group 2000). However it is worth noting that the DYT3 variant, which is X-linked and affects mostly Filipino men and rarely women (Evidente 2005), causes dystonia of varying severity but, importantly, is also characterized by parkinsonism. This observation strengthens the aforementioned association between PD, chromosome X, and male neurodegenerative disorders.
Psychiatric Disorders
La Tourette syndrome and autism are mental disorders with marked male prevalence (9:1 and 4:1, respectively) (Bao and Swaab 2010) whose nature is inherently complex. Genetics seem to have a critical role in the pathogenesis of Tourette syndrome; cytogenetic and linkage analyses have uncovered a number of loci and several genetic mutations but the disorder may occur as the result of complex interactions between environmental and genetic factors (Deng and others 2012). The syndrome seems to have, at least in part, an autosomal dominant pattern of inheritance, but interestingly, all the genes identified seem to have mainly a neuronal function (Deng and others 2012). A X-linked gene (neuroligin-4) has been identified (Lawson-Yuen and others 2008), and the larger frequency in males than in females has been explained by a X-link modified model for the disease (Comings and Comings 1986).
The genetics of autism is similarly baffling. Many cases of autism appear to be caused by several abnormal genes acting in concert, and most of the chromosomes have been implicated in the genesis of autism. However, aberrations on the long arm of chromosome 15 and numerical and structural abnormalities of the sex chromosomes have been most frequently reported (Gillberg 1998; Jamain and others 2003).
In more general terms, there is strong evidence of sexual dimorphism in the expression of disorders characterized by abnormalities in attention and/or impulsivity that include autism but also attention deficit hyperactivity disorder (ADHD) and addiction (Trent and Davies 2012). There is a substantial male prevalence in ADHD (between 3:1 and 9:1 but this may decrease with age) (Swanson and others 1998). Interestingly though, sex bias appears to be subtype specific with the inattentive type more prevalent in girls, and boys being more frequently diagnosed with the hyperactive-impulsive and combined subtypes (Biederman and others 2002; Lahey and others 1994). Hence, different cell aetiology in the attention/impulsivity systems may account for sex-specific presentations. Substantial evidence supports a role for the sex-linked genes SRY, STS, and MAO in attention and impulsivity (Trent and Davies 2012). Monoamine oxidase A (MAO-A) catalyzes the oxidative deamination of amines, including serotonin, norepinephrine, and dopamine. The MAO-A gene, located on X close to the MAO-B gene, is modulated by SRY (Wu and others 2009) and has been associated with vulnerability to ADHD (Gizer and others 2009) and autism (Cohen and others 2003; Davis and others 2008; Yoo and others 2009) as well as the so-called “Brunner Syndrome,” a rare condition first identified in the male members of a Dutch family characterized by aggression and mental retardation where MAO-A activity is markedly reduced (Brunner and others 1993). MAO-A is most abundant in noradrenergic neurons, is moderately contained by serotoninergic neurons, and present in lower levels in dopaminergic neurons while MAO-B is expressed in both neurons and glia (Luque and others 1995).
Schizophrenia is a mental disorder with an increased male risk (Bao and Swaab 2010). It is distinct from X-linked mental retardations although chromosome X abnormalities and sex-specific patterns of transmission have been identified (Crow 2013; Goldstein and others 2011). Its aetiology is unknown but the proposed models, whether they involve striatal hyper-dopaminergia (Howes and Kapur 2009) or diminished glutamatergic transmission or an interaction of the two (Lisman and others 2008), all point to neurons, and particularly parvalbumin positive interneurons, as the cell conduit of the disorder (Jiang and others 2013; Marin 2012). However, it is of note that in the late-onset patients the observed male-to-female ratio reverts with a higher proportion of elderly female patients (Meesters and others 2012). Here the speculative application of the X-linked hypothesis would propose a heterogeneous aetiology across ages of onset with a prevalence of non-neuronal pathology in the late stage of the disease. This is supported by tissue findings that point to gliosis as the pathological hallmark of this population subtype (Nasrallah and others 1983).
Finally, prevalence is reversed in affective disorders as women are more vulnerable than men to disorders that affect emotions such as major depression, generalized anxiety disorders, panic disorders, posttraumatic stress disorders, and phobias (Holden 2005). The higher risk of the female population in this context has been generally associated to a number of environmental/social stressors; here it suggests a non-neuronal aetiology. In this context, the X-linked hypothesis could be used to support the validity of animal models that do not have a modified neuronal substrate as cell conduit for the simulation of the disorder but where, for example, glial or immune cells play the central role in the modulation of the brain response (Quesseveur and others 2013; Yirmiya 1996).
Conclusion
The X-linked hypothesis presented here supports the general idea, already highlighted by others (Cahill 2006), that gender is an important interacting factor in the study of brain disorders. In particular, it formalizes the seemingly persistent association between the male gender and neuronal dysfunction across brain disorders.
Note that this hypothesis does not preclude any further modulation of brain disorders by the sexual genes. For example, consider Leber’s hereditary optic neuropathy that is another degenerative disorder of neuronal origin that overwhelmingly affects males (Stys and others 2012). Leber’s neuropathy results from mutations in the mitochondrial genome that in humans is inherited via the maternal lineage. As this disorder has a marked male prevalence, a role for nuclear genes on chromosome X has long been pursued with uncertain results (Chalmers and others 1996; Hudson and others 2005; Shankar and others 2008; Vikki and others 1991). Recently, however, Giordano and coworkers (2011) have shown that the apparent X-link with neural function in this disease can be explained by the ameliorating effect of estrogens on mitochondrial dysfunction. Mitochondrial DNA codes for 37 genes that are essential for mitochondrial function, 13 of which provide instructions for making enzymes involved in oxidative phosphorylation; in the human brain, selective populations of neurons have been the ones showing the highest sensitivity to defects in the mitochondrial respiratory chain (Betarbet and others 2000) and the action of female hormones seem to lessen the disease burden (Giordano and others 2011).
In the wider context of gender resilience/susceptibility to disease, recent work from Chen and colleagues (2013) has put forward an alternative and interesting perspective on male vulnerability and neurodevelopmental variation; they have shown that SRY, the gene that ultimately defines the maleness of a human embryo by initiating the genetic program that leads to the formation of testes and fetal testosterone, appears to be unreliable. More generally this finding supports the view that male sexual development is less stable than other genetic programs.
Another recent study from Trabuzni and colleagues (2013) has demonstrated major sex differences in gene expression and splicing in the adult human brain mRNA that were most represented by X-linked genes in the medulla and subcortical nuclei (substantia nigra, hippocampus, putamen), the cerebellum, and the temporal and frontal cortices. This finding could potentially indicate spatial heterogeneity in the gender associated risk that we have here espoused.
The X-linked hypothesis is therefore proposed as a useful conceptual tool in the quest for the aetiology of complex brain disorders; ultimately, the hypothesis could be put fruitfully to use by guiding the development of biomarkers sensitive to early signs of degeneration. In vivo markers able to assay the general health of the individual brain cell types (e.g., neuronal subtypes, astrocytes, microglia, oligodendrocytes) could be applied to studies of longitudinal trajectories of brain development and change. Such studies should be enriched with populations at risk and be powered under the assumption of a significant heterogeneity in the clinical cohorts. Positron emission tomography and magnetic resonance imaging are already able to provide some of these markers or at least good proxies.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work has been funded in part by the PET Methodology Programme Grant from the Medical Research Council UK (G1100809/2).