
Abstract
It is generally accepted that proper activation of N-methyl-
Introduction
N-methyl-
NMDARs are targeted to both synaptic and extrasynaptic sites. Synaptic NMDARs (syn-NMDARs) are located on the plasma membrane within 200 to 300 nm of the post-synaptic density (PSD). The extrasynaptic NMDARs (ex-NMDARs) are located on spine necks, dendritic shafts, or somas, which are further away from the PSD. Syn-NMDARs are activated by tonic and activity-dependent glutamate release from the presynaptic terminal. While certain populations of the ex-NMDARs are constitutively activated by ambient glutamate and contributes to the tonic currents (Le Meur and others 2007; Papouin and others 2012; Sah and others 1989), possibly the majority of ex-NMDARs are activated by glutamate spillover following intense synaptic activity or massive ectopic glutamate release following insults and brain trauma (Harris and Pettit 2008; Rossi and others 2007) (Fig. 1). As emerging proposals advocate that attenuation of excitotoxicity may be better achieved by targeting specific sub-populations of NMDARs rather than general inhibition, this review focuses on the function of syn- and ex-NMDARs in cell fate determination.
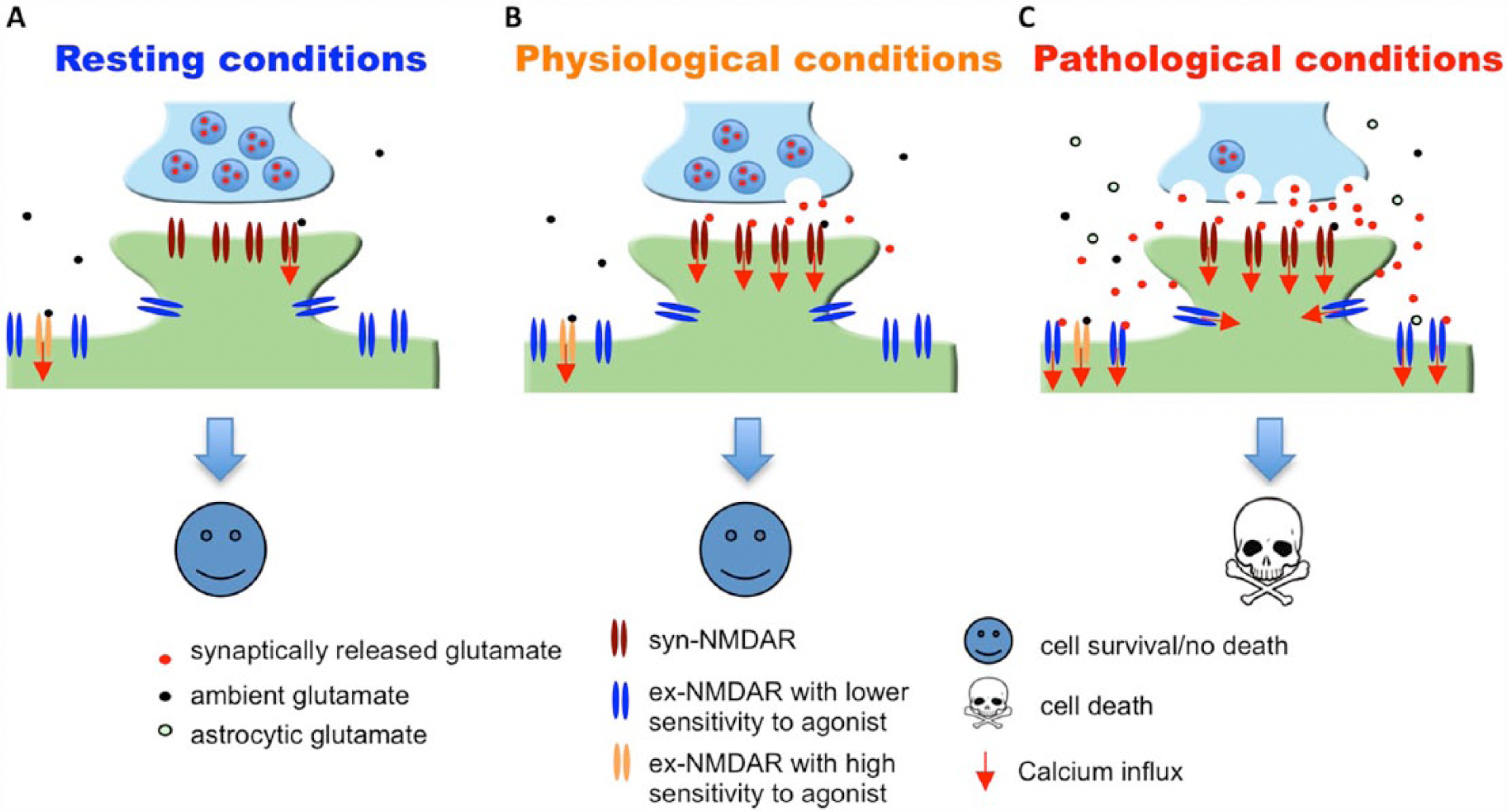
Massive and prolonged co-activation of synaptic (syn)-NMDARs and extrasynaptic (ex)-NMDARs leads to cell death. (A) It is estimated that the EC50 of glutamate to activate naïve as well as recombinant NMDAR is 2 to 4 µM, and that the ambient extracellular glutamate concentration is between 0.5 to 5 µM. Thus, under resting conditions, ambient glutamate may be sufficient to activate certain population of syn-NMDARs as well as ex-NMDARs. (B) Under physiological conditions, glutamate released from the presynaptic terminal activates syn-NMDARs. (C) Under pathological conditions, massive release of glutamate from both neuronal terminal and astrocytes leads to significant co-activation of syn- and ex-NMDARs. Short-term co-activation, which may occur during intensive brain activity and brief insult, does not necessarily lead to significant cell death. If the magnitude of receptor co-activation is correlated to the degree of cell death, inhibition of syn- or ex-NMDARs or partial co-inhibition will attenuate neurodegeneration. The existence of ex-NMDARs showing either high or low sensitivity to receptor agonist is suggested by that certain ex-NMDARs are activated by ambient glutamate (Le Meur and others 2007; Sah and others 1989) and certain ex-NMDARs are only activated by high level NMDA (Zhou and others 2013b). NMDAR = N-methyl-
The Role of Syn- and Ex-NMDARs in Cell Death following Pathological Insults
It is known that the activation of NMDARs triggered by synaptic activities is required for normal brain function. Pharmacological manipulations that only activate syn-NMDARs are protective rather than causing cell death. While the tonic and constitutive activation of NMDARs in healthy brains (Le Meur and others 2007; Sah and others 1989) is maintained by ambient extracellular glutamate (at concentrations ranging from 0.5 to 5 µM) (Featherstone and Shippy 2008), glutamate levels exceeding 20 µM start to cause excitotoxicity. As the physiological activity–triggered presynaptic release results in glutamate level in the synaptic cleft reaching 1000 µM (Featherstone and Shippy 2008), exposure of syn-NMDARs alone to high concentration of agonists is unlikely to cause death (Fig. 1).
It has been confirmed by many research groups that low levels of glutamate (e.g., 10-20 µM) or NMDA (e.g., 10-20 µM) do not cause death in cultured neurons (Chandler and others 2001; Zhou and others 2013b). Increasing concentrations progressively cause incremented death, and the maximal death level can be observed after exposure to 50 to 100 µM NMDA (Zhou and others 2013b). These phenomena have raised several important questions. Does low agonist concentration only activate syn-NMDAR or ex-NMDAR or partially activate both? Does higher agonist concentration preferentially activate more syn-NMDAR or more ex-NMDAR or both? Is cell death triggered by high concentrations of agonist caused by the overactivation of syn-NMDAR or ex-NMDAR or the co-activation of both?
The prevailing theory, which is supported by data collected from many labs, emphasizes that glutamate excitotoxicity is predominantly regulated by ex-NMDARs. In a seminal study, Hardingham and colleagues found that the activation of syn-NMDARs by bicuculline treatment activates the pro-survival molecule CREB. Subsequent bath incubation with high concentration glutamate shuts off CREB signaling and causes severe neuronal death (Hardingham and others 2002). Although bath glutamate incubation likely activates both syn- and ex-NMDARs (see more detailed discussion later), the authors suggest that the activation of ex-NMDARs counteracts syn-NMDARs and suppresses pro-survival signaling. Inspired by this interesting initial study, numerous follow-up investigations have tried to isolate the ex-NMDARs and examined their role in cell death. To specifically block syn-NMDARs, neurons were first treated with bicuculline along with the irreversible use-dependent NMDAR antagonist MK-801. Hence, the subsequent bath application of NMDA or glutamate would only activate ex-NMDARs. It has been demonstrated that ex-NMDAR activation leads to significant death in neurons cultured from multiple different brain regions (for a review, see Hardingham and Bading 2010).
However, the “ex-NMDAR theory” of excitotoxicity has limitations in explaining some of the existing phenomena. First, it is known that ex-NMDARs are dominant receptors in young developing neurons, which are resistant to glutamate and NMDA insults (Hardingham and Bading 2002; Friedman and Segal 2010; Zhou and others 2009). Second, glutamate excitotoxicity is absent in retinal ganglion cells, in which there is no syn-NMDAR expression, and the NMDAR current is only mediated by ex-NMDARs (Chen and Diamond 2002; Ullian and others 2004). Thus, one can argue that the activation of ex-NMDARs alone is not sufficient to cause death (Fig. 2).
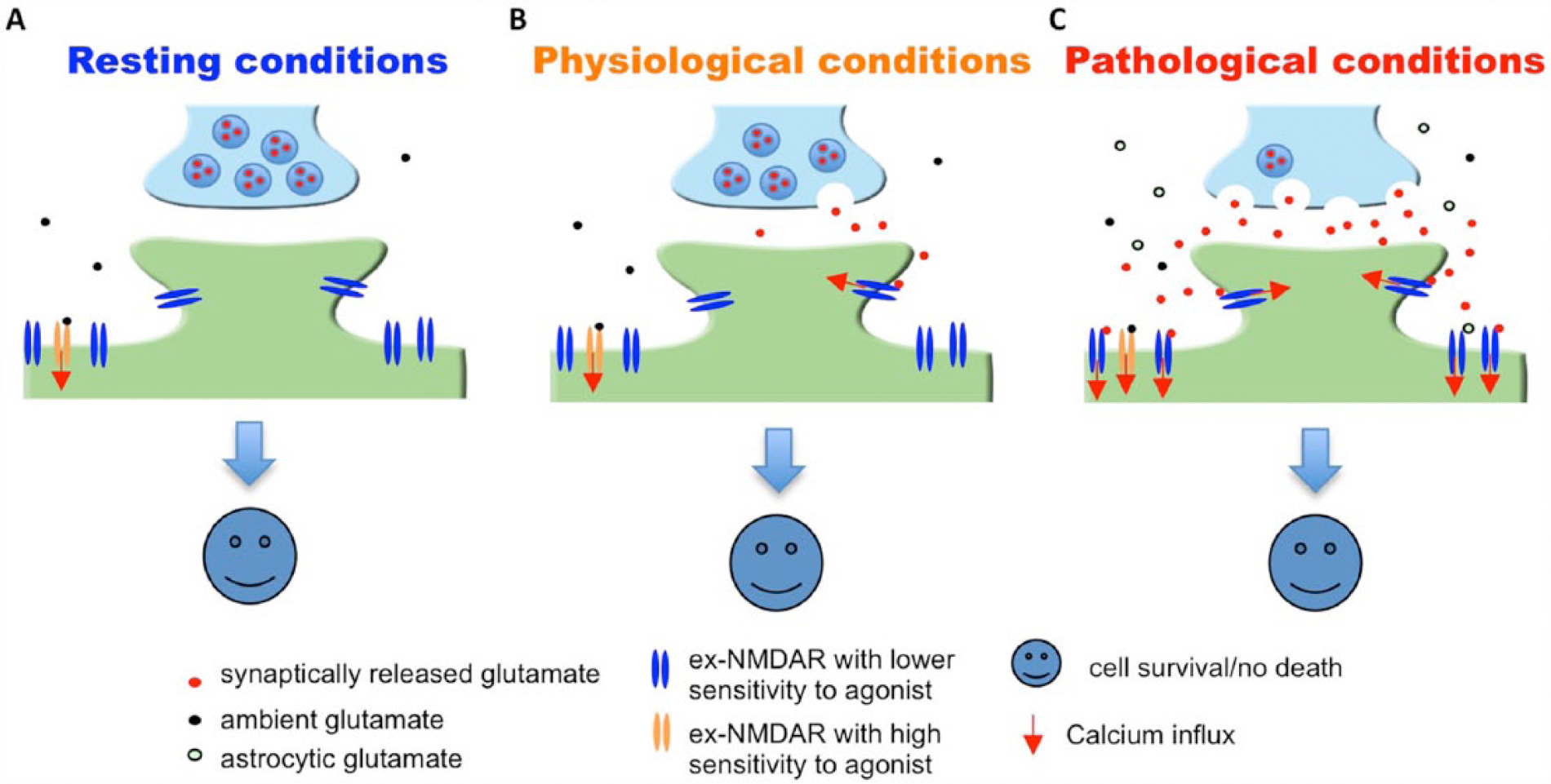
Activation of ex-NMDARs in young or retinal ganglion neurons does not lead to cell death. It is known that NMDAR function is mainly mediated through extrasynaptic (ex)-NMDARs in young developing neurons and retinal ganglion neurons. It is known that, in these neurons, the activation of ex-NMDARs by ambient glutamate (A), synaptically released glutamate (B), or massive glutamate release from both synaptic terminals and astrocytes (C) does not trigger cell death. NMDAR = N-methyl-
Intriguingly, several studies suggest the function of syn-NMDARs in excitotoxicity. Sattler et al. showed that decreasing the number of syn-NMDARs dampens the oxygen glucose deprivation (OGD)-induced cell death (Sattler and others 2000). By blocking syn-NMDARs with MK-801 or degrading ambient glutamate with glutamate pyruvate transaminase, a recent study demonstrated that syn-NMDARs participate in hypoxic excitotoxicity (Wroge and others 2012). Notably, Papouin and others (2012) discovered that syn-NMDARs and ex-NMDARs in hippocampal neurons are differentially gated by the endogenous coagonist
By using cultured cortical neurons, Zhou and others (2013b) confirmed that the activation of syn-NMDARs does not cause cell death. Along with the syn-NMDAR activation, increasing amount of ex-NMDAR activation correlated with an increasing degree of cell death and the disruption of intracellular Ca2+ homeostasis. After blocking syn-NMDARs, high concentrations of NMDA progressively activated an increasing amount of ex-NMDARs, but failed to trigger cell death and the disruption of intracellular Ca2+ homeostasis. Blocking syn-NMDARs also totally rescued OGD-induced cell death. The results demonstrate that the activation of either syn-NMDARs or ex-NMDARs alone is not sufficient to cause excitotoxicity. This work suggests that cell death depends on the co-activation of NMDARs at both synaptic and extrasynaptic sites (Fig. 1). Hence, blocking either syn-NMDAR (Sattler and others 2000; Papouin and others 2012; Wroge and others 2012; Zhou and others 2013b) or ex-NMDAR (Tu and others 2010) attenuates excitotoxicity.
Role of Ex-NMDARs in Mediating Physiological Functions
The “ex-NMDAR theory” on excitotoxicity is not consistent with the fact that the ex-NMDARs are also involved in physiological functions of the brain. Prior to synapse formation, the activation of ex-NMDARs is crucial for neuronal development, such as synaptogenesis, neuritogenesis, and neuronal migration and differentiation. Furthermore, it has been demonstrated that the activation of ex-NMDARs by glutamate spillover leads to the initiation of NMDA spikes in basal dendrites, which is crucial for the integration of synaptic input (Chalifoux and Carter 2011; Oikonomou and others 2012). As indicated by the studies of Fellin and others (2004) and Angulo and others (2004), astrocytic glutamate-triggered ex-NMDAR activation causes synchronous firing, which is suggested as a fundamental element in information processing. Moreover, 25- to 200-Hz short bursts, which occur during exploratory behavior, are sufficient to activate ex-NMDARs. The authors suggest that cross-talk between synaptic and extrasynaptic receptors increases synaptic strength (Harris and Pettit 2008). Some recent studies demonstrate that ex-NMDARs play a major role in long-term depression (LTD), a cellular substrate of neuroplasticity (Papouin and others 2012; Liu and others 2013). In the visual cortex, the expression of ex-NMDAR is up-regulated following sensory stimulation. Such activity-dependent changes of ex-NMDAR are thought to prime synapses for potentiation/strengthening (Eckert and others 2013). There is also evidence to support the notion that the activation of ex-NMDARs by back-propagating action potentials regulates dendritic excitability through down-regulation of h-channel conductance (Wu and others 2012). In addition to synaptic glutamate spillover, glutamate exocytosis from astrocytes also activates ex-NMDARs and controls synaptic activity and strength (Jourdain and others 2007).
It is important to note that ex-NMDAR activation may be accompanied by syn-NMDAR activation, regardless of whether it is in physiological or pathological conditions (Fig. 1). The lack of excitotoxicity following physiological co-activation of syn-NMDARs and ex-NMDARs may be due to the short duration and low degree of the co-activation. An in vitro study demonstrates that brief receptor co-activation (e.g., less than 4-minute exposure to toxic levels of NMDA) leads to the up-regulation of pro-survival rather than apoptotic signaling. Consistently, a very brief ischemic insult can be neuroprotective (Zhou and others 2013b). Although the concentration of ambient extracellular glutamate is low in healthy brains, it is sufficient to cause tonic activation of NMDARs at the extrasynaptic locations (Le Meur and others 2007; Papouin and others 2012; Sah and others 1989). This suggests that even chronic constitutive activation of ex-NMDARs (presumably at low level though) is not neurotoxic.
Pharmacological Differences between Syn-NMDAR and Ex-NMDAR
The physiological and pathological functions of syn- and ex-NMDAR may be better understood by examining the effects of specific inhibitors. As the co-activation of both receptors is required to trigger excitotoxicity, specific inhibition of the ex-NMDARs may offer favorable therapeutic effects to suppress NMDAR overactivation without dampening synaptic function. Among the available NMDAR antagonists, memantine has been used for the treatment of Alzheimer’s disease, and suggested to preferentially block ex-NMDARs (Xia and others 2010). However, Wroge and others (2012) found that memantine blocks EPSC mediated by either syn- or ex-NMDARs. Further, intracellular signaling triggered by either synaptic or extrasynaptic activation is suppressed by memantine (Zhou and others 2013b). Consistent with the notion that co-activation of both receptors is required for excitotoxicity, partial and simultaneous blockade of syn- and ex-NMDARs by low-dose memantine suppresses NMDA-induced cell death (Zhou and others 2013b). The non-specific effects are also suggested by the observation that memantine attenuates the synaptic NMDAR-mediated LTP (Frankiewicz and others 1996; Papouin and others 2012) and the extrasynaptic NMDAR-mediated LTD (Liu and others 2013; Papouin and others 2012; Scott-McKean and Costa 2011).
Better understanding of pharmacological and structural differences between syn- and ex-NMDAR may aid the development of specific inhibitors. Previous studies have suggested certain factors that may differentially affect the channel and pharmacological properties of synaptic and extrasynaptic receptors. The difference in channel properties may be due to a different density and component of scaffolding proteins that anchor NMDARs to dendritic spines and shafts (Gladding and Raymond 2011). The enrichment of NR3A subunits (Barria and Malinow 2002; Perez-Otano and others 2006), as well as specific splice variants and phosphorylation (Goebel-Goody and others 2009; Li and others 2002) in the ex-NMDARs may also render different agonist and co-agonist sensitivity from that of syn-NMDARs. Notably, it has been demonstrated that the ratio of synaptic to extrasynaptic NMDARs undergoes significant changes throughout neural development, partially due to the expression switches between NR2A and NR2B. Some studies suggest that NR2A and NR2B regulate synaptic and extrasynaptic function as well as LTP and LTD, respectively. However, recent work suggests that NR2A and NR2B are present in both syn- and ex-NMDARs, and are involved in regulating intracellular signaling mediated by either syn- or ex-NMDARs (Zhou and others, 2013a). Interestingly, Papouin and others (2012) have found that the syn-NMDARs are gated by co-agonist
It is estimated that the EC50 of glutamate to activate the NMDARs is 2 to 4 µM. Glutamate at ~50 µM triggers maximal response. As high but not low concentrations of NMDAR agonists are excitotoxic, the current understanding predicts that there may be at least two populations of ex-NMDARs. One is sensitive to low level and ambient agonist, and responsible for tonic and constitutive NMDAR current (Le Meur and others 2007). The other is only activated by high level NMDA or glutamate, which may occur transiently in physiological conditions and chronically in neurodegenerative situations. By using fluorescence-based imaging, Zhou and others (2013b) assessed the NMDAR-mediated Ca2+ influx in live neurons. Neurons were first pretreated with bicuculline and MK801 to block the synaptic receptors. MK801 in the pretreatment cocktail should have also irreversibly blocked the active ex-NMDARs that mediate the tonic currents. The subsequent application of 15 µM NMDA failed to induce Ca2+ influx, but a higher level of NMDA (from 20 to 50 µM) did. In another experiment, neurons were pre-treated with 15 µM NMDA and MK801, so that receptors sensitive to NMDA at ≤15 µM should be blocked. Subsequent application of bicuculline or 15 µM NMDA failed to cause Ca2+ influx, but 20 to 50 µM NMDA did. This indicates that the majority of the ex-NMDAR is sensitive to higher NMDA (i.e., >15 µM), and low NMDA (at ≤15 µM) activates most of the syn-NMDARs. Based on this finding, we propose a simple high-throughput strategy to screen for synaptic and extrasynaptic NMDAR-specific inhibitors (Fig. 3).
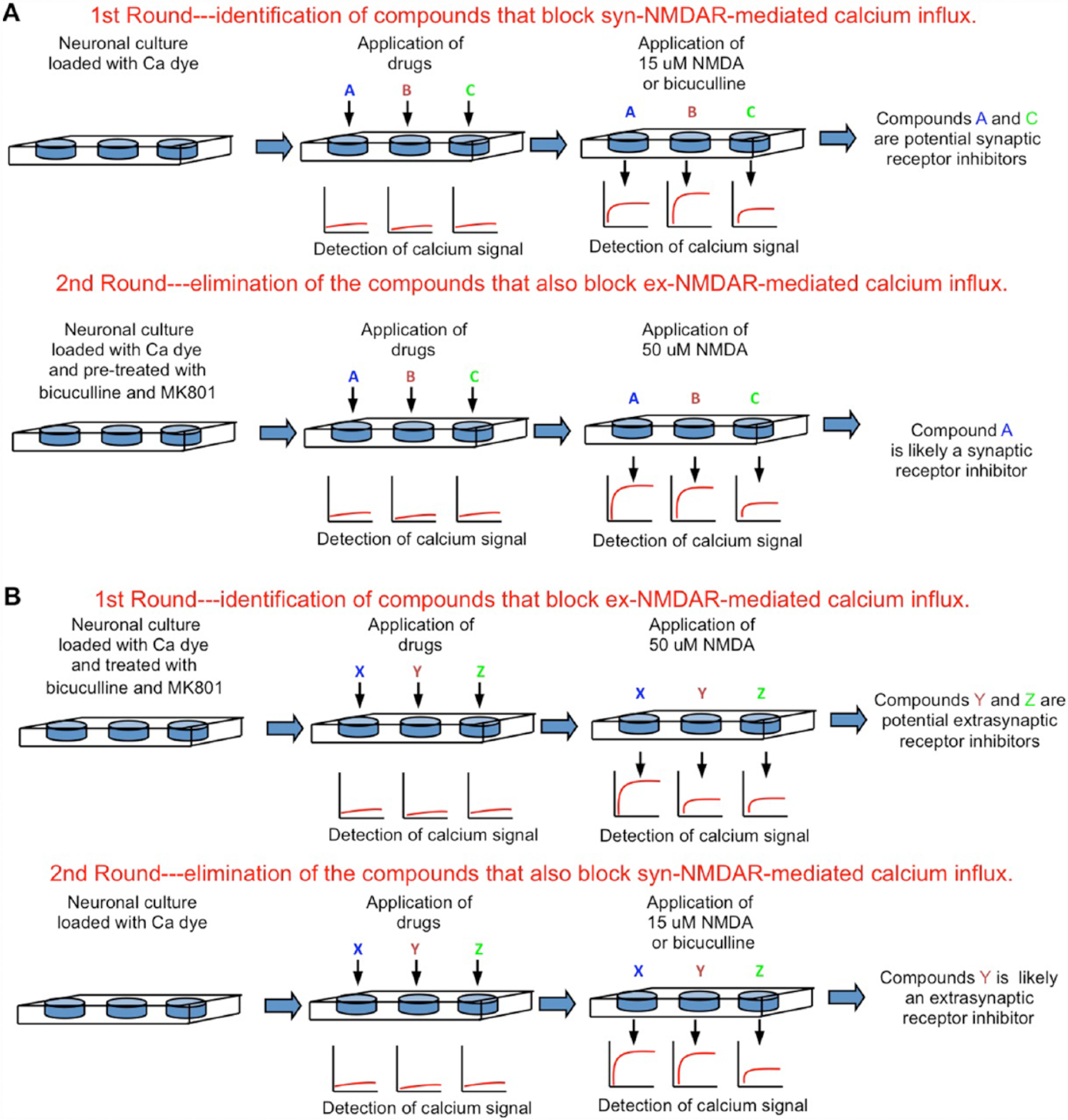
High-throughput screening strategies to identify inhibitors for synaptic or extrasynaptic NMDARs. Primary cultured neurons are seeded in 96- or 384-well plates. The NMDAR-mediated Ca2+ influx will be detected by an automated plate reader using fluorescent Ca2+-sensitive dyes. (A) Strategy to screen for synaptic receptor inhibitors. (B) Strategy to screen for extrasynaptic receptor inhibitors. NMDAR = N-methyl-
The existence of ex-NMDARs with high and low agonist sensitivity may help explain the different cell fate in normal and degenerative brains. In ischemic stroke, massive glutamate release elevates extracellular concentration and may in turn activates ex-NMDARs that are not sensitive to low ambient glutamate. The low-sensitivity ex-NMDARs that are only sensitive to high level but not ambient level agonist may also be transiently activated by significant glutamate spillover during exploratory behavior in rats when bursts of synaptic transmission are detected (Harris and Pettit 2008). In the brains of Alzheimer’s patients or animal models, there is a reduction of glutamate transporters, reduced glutamate uptake, and therefore an increase in ambient glutamate level, which is hypothesized to activate more ex-NMDARs (Parsons and Raymond 2014).
The higher agonist sensitivity in syn-NMDARs (than the “pathological” ex-NMDARs) may offer an explanation for the dose-dependent cell death induced by bath NMDA application. In our opinion, bath incubation does not always trigger global NMDAR activation. Low NMDA (e.g., 15 ≤µM) mainly activates synaptic NMDAR, and hence can be neuroprotective rather than neurotoxic. Most of the “pathological” ex-NMDARs are only activated by higher levels of NMDA (i.e., >20 µM). Thus, high but not low NMDA triggers receptor co-activation and causes excitotoxicity. Other evidence also supports the idea that syn-NMDARs may be more sensitive to agonist than ex-NMDARs. While promoting glutamate efflux from a small population of astrocytes in neuron/glia co-cultures mainly activates syn-NMDARs, glutamate efflux from significantly more astrocytes causes ex-NMDAR activation (Gouix and others 2009).
Concluding Remarks and Future Directions
Accumulating efforts are elucidating the role of syn- and ex-NMDARs in physiological and pathological functions. Although elevated ex-NMDAR activity is associated with neurodegeneration, analysis of the existing data suggests that the activation of ex-NMDAR alone may not lead to significant excitotoxicity. However, preferential inhibition of the overactivated ex-NMDARs may represent a favorable approach to prevention of neurodegeneration. Specific and potent inhibitors targeting the ex-NMDARs are not yet available. The development of effective high-throughput screening strategies would move us toward that goal.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by start-up fund from Nanjing Medical University affiliated Changzhou Hospital (to XZ), Science and Technology Developmental Key Project of Nanjing Medical University (2013NJU212 to XZ), Changzhou Applied Basic Research Program (CJ20130023 to ZC), NIH grants (MH093445 and NS072668 to HW), and American Heart Association postdoctoral fellowship (10POST4550000 to XZ).