
Abstract
The small GTP-binding protein Rho plays an important role in several cellular functions. RhoA, which is a member of the Rho family, initiates cellular processes that act on its direct downstream effector Rho-associated kinase (ROCK). ROCK inhibition protects against dopaminergic cell death induced by dopaminergic neurotoxins. It has been suggested that ROCK inhibition activates neuroprotective survival cascades in dopaminergic neurons. Axon-stabilizing effects in damaged neurons may represent another mechanism of neuroprotection of dopaminergic neurons by ROCK inhibition. However, it has been shown that microglial cells play a crucial role in neuroprotection by ROCK inhibition and that activation of microglial ROCK mediates major components of the microglial inflammatory response. Additional mechanisms such as interaction with autophagy may also contribute to the neuroprotective effects of ROCK inhibition. Interestingly, ROCK interacts with several brain factors that play a major role in dopaminergic neuron vulnerability such as NADPH-oxidase, angiotensin, and estrogen. ROCK inhibition may provide a new neuroprotective strategy for Parkinson’s disease. This is of particular interest because ROCK inhibitors are currently used against vascular diseases in clinical practice. However, it is necessary to develop more potent and selective ROCK inhibitors to reduce side effects and enhance the efficacy.
Introduction
The small GTP-binding protein Rho plays an important role in several cellular functions. RhoA, which is a member of the Rho family, initiates cellular processes that act on its direct downstream effector Rho-associated kinase (ROCK) (Katoh and others 1998; Zhang and others 2007). Two isoforms encoded by two different ROCK genes have been described: ROCK I and ROCK II (Nakagawa and others 1996). ROCK II (also known as ROCKα) is preferentially expressed in the brain. Abnormal activation of the RhoA/ROCK pathway is known to be involved in cardiovascular diseases, oncology, pulmonary, and renal diseases, and ROCK inhibitors have been suggested as a promising therapeutic strategy (Mueller and others 2005; Olson 2008; Pan and others 2013). In the central nervous system, abnormal activation of the RhoA/ROCK pathway has been observed in models of spinal cord injury, inflammatory demyelinating diseases, stroke, Alzheimer’s disease and other diseases (Chen and others 2013; Herskowitz and others 2013; Mueller and others 2005; Sheikh and others 2009). This has been related to the involvement of ROCK in neuroinflammatory processes that play a major role in these diseases (Greenwood and others 2003; Sheikh and others 2009; Walters and others 2002). Furthermore, ROCK activation is also known to be involved in axonal collapse and retraction, and ROCK inhibition may also induce therapeutic effects by axon-stabilizing functions and promotion of neurite outgrowth (Gallo 2004; Katoh and others 1998; Lehmann and others 1999; Lingor and others 2007). ROCK inhibition has also been observed to induce antiapoptotic effects and beneficial influence on neuron survival in several models of brain diseases such as Huntington disease (Bowerman and others 2012), amyotrophic lateral sclerosis (Tönges and others 2014), and retinal degeneration (Bermel and others 2009; Koch and others 2014).
Both ROCK I and ROCK II phosphorylate a variety of protein substrates at serine or threonine residues, and more than 20 ROCK substrates have been identified. The first characterized targets of ROCK were the myosin light chain (MLC) and the myosin binding subunit of the MLC phosphatase, and most of the ROCK substrates are cellular proteins associated with the regulation of actin cytoskeleton (see Amano and others 2010; Amin and others 2013). ROCK has also been recognized as a major regulator of the morphological events that occur during the execution phase of apoptosis, including cell contraction, dynamic membrane blebbing, and fragmentation of apoptotic cells (Shi and Wei 2007), and several ROCK substrates are involved in the regulation of cell death and survival, which include phosphatase and tensin homologue (PTEN), Akt, Bcl-2, GSK-3β, and several others. Major ROCK substrates and effects mediated by the RhoA/ROCK pathway have been summarized in Figure 1. A detailed analysis of all potential ROCK targets is beyond the scope of the present article. Several potential ROCK targets that have been shown to be involved in dopaminergic neuron degeneration will be briefly analyzed, and the review will be focused on targets and mechanisms that have been demonstrated to mediate ROCK-induced dopaminergic degeneration and the neuroprotective effects of ROCK inhibition in different models of Parkinson’s disease (PD).
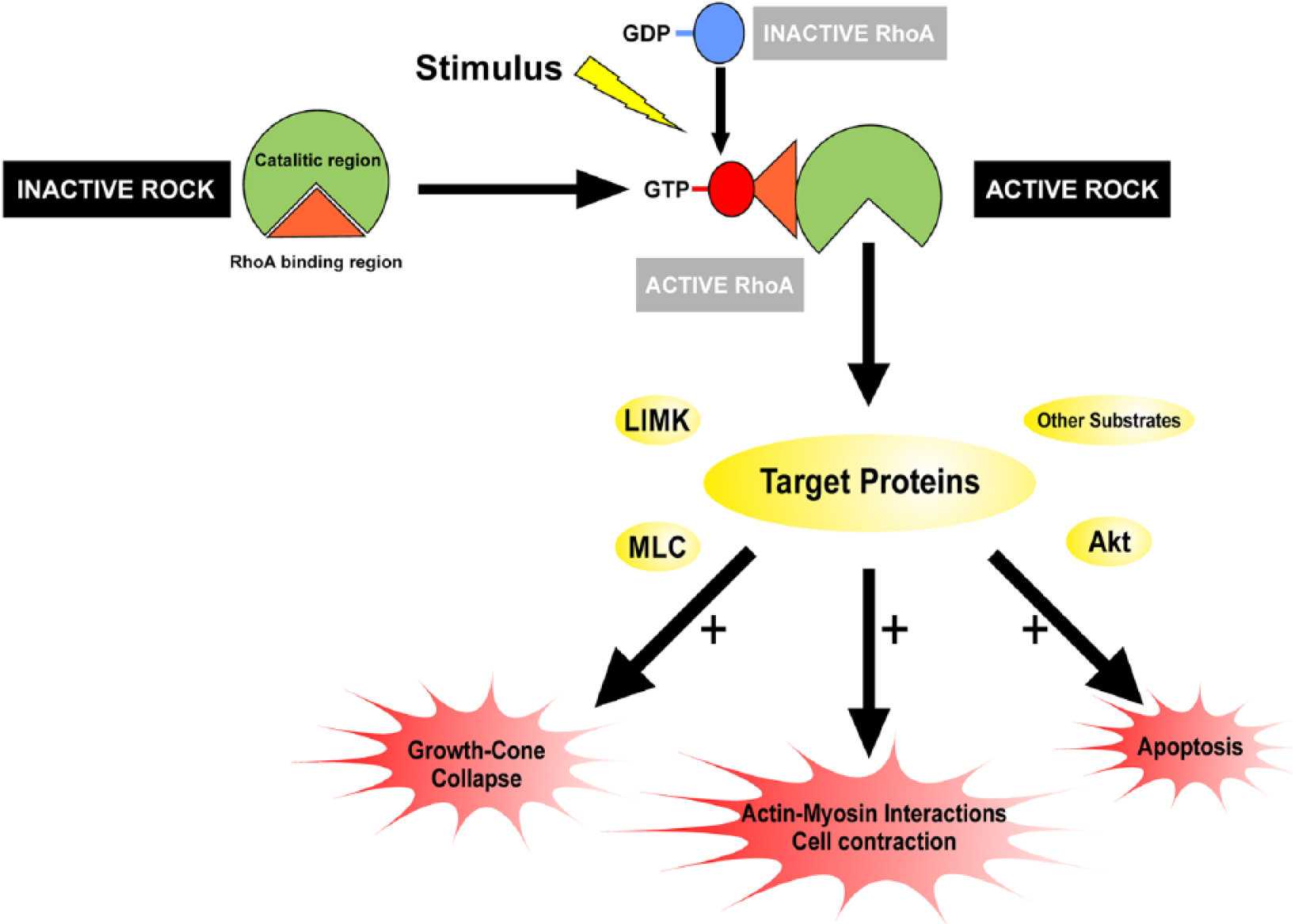
Schematic drawing of the effects mediated by the RhoA/ROCK Pathway. Activated RhoA binds to the Rho binding region of ROCK in response to stimuli, thereby leading to ROCK activation. Activated ROCK acts on several direct or indirect target proteins such as LIM domain kinase (LIMK), myosin light chain (MLC), myosin light chain phosphatase (MLCP), collapsin response mediator protein 2 (CRMP2), phosphatase and tensin homologue (PTEN), and Akt, and leads to actin cytoskeleton rearrangement and regulation of apoptotic events. For details, see Amano and others (2010), Amin and others (2013), and Shi and Wei (2007). GDP = guanosine diphosphate; GTP = guanosine triphosphate.
The pathogenic mechanism of PD appears to be multifactorial. It has been shown that several genes are mutated or deleted in familial PD. However, the etiology of sporadic, idiopathic PD, which accounts for most cases of PD, is still unclear. A number of mechanisms have been shown to be involved in dopaminergic neuron degeneration in PD, including mitochondrial dysfunction, oxidative stress, environmental toxins, and impairment of the ubiquitin-proteosome system (Goldman 2014; Gonzalez-Hernandez and others 2010; Jellinger 2013; Olanow 2007; Sanchez-Iglesias and other 2009; Tolleson and Fang 2013). Although neuroinflammation was initially considered to be a simple consequence of neuronal degeneration, it is now clear that it plays a major role in the progression of dopaminergic cell death, even though it is unlikely to be a primary cause of PD (Hirsch and others 2012; Hoban and others 2013; Lindqvist and others 2013). These pathogenic factors are not mutually exclusive, and one of the key aims of current PD research is to discover the mechanisms involved in possible interactions between these pathways, which result in dopaminergic neuron degeneration. Interestingly, several major mechanisms related to ROCK activation play a major role in dopaminergic degeneration and may be counteracted by ROCK inhibition. First, neuroinflammation plays a major role in the progression of dopaminergic cell death; a marked microglial reaction has been observed in the nigra and striatum of brains from PD patients and PD animal models (Gerhard and others 2006; Ouchi and others 2005; Rodriguez-Pallares and others 2007). Second, initial axon degeneration followed by dopaminergic neuron degeneration has also been suggested to occur in PD, in a “dying back” model of the disease (Burke and O’Malley 2013; Chu and others 2012). Third, apoptotic processes play a major role in dopaminergic cell death (Alves da Costa and Checler 2011; Levy and others 2009). Finally, disruption of autophagy in dopaminergic neurons is also associated with progression of the pathology of PD (Dehay and others 2010), and ROCK activation is involved in the autophagic process (Bauer and Nukina 2009).
Rho-Kinase Activation and Dopaminergic Neuron Degeneration
Over the last few years, several studies have observed that ROCK inhibition protects against dopaminergic cell death induced by the dopaminergic neurotoxin MPTP/MPP+ in mice and in cultures of dopaminergic neurons (Barcia and others 2012; Borrajo and others 2014b; Tönges and others 2012; Villar-Cheda and others 2012) (Fig. 2). In mice, treatment with MPTP induced a loss of dopaminergic neurons in the substantia nigra compacta (SNc) and striatal dopaminergic terminals, and this loss was significantly reduced by simultaneous treatment with ROCK inhibitors (i.e., fasudil or Y-27632; Barcia and others 2012; Tönges and others 2012; Villar-Cheda and others 2012). Consistent with this, the MPTP-induced loss of striatal dopamine and motor impairment were significantly reduced by treatment with ROCK inhibitors (Tönges and others 2012). Furthermore, it has also been observed that MPTP induces a significant increase in activation of the RhoA/ROCK II pathway in the mouse SNc (Villar-Cheda and others 2012). Interestingly, significant increases in the expression of RhoA and ROCK II mRNA levels and in ROCK activity were observed soon after the first MPTP injection (i.e., prior to any significant loss of neurons; Jackson-Lewis and others 1995; Wu and others 2003), which suggests that the increase in ROCK activity is involved in dopaminergic degeneration rather than being a consequence of the loss of dopaminergic neurons in the SNc. Increased activation of the RhoA/ROCK II pathway was still apparent when the degeneration process was almost completed (i.e., 7 days after the final MPTP injection; Villar-Cheda and others 2012). In primary neuron-glia mesencephalic cultures, ROCK inhibitors also provided neuroprotection against MPP+-induced dopaminergic neuron degeneration (Borrajo and others 2014b; Tönges and others 2012; Villar-Cheda and others 2012). Confirmation of the neuroprotective effects of ROCK inhibitors in cultures is of interest as it shows that neuroprotection is not merely a consequence of changes in brain vessel function (see below). Furthermore, the MPTP-induced increase in ROCK activity has also been confirmed in primary cultures (Villar-Cheda and others 2012).
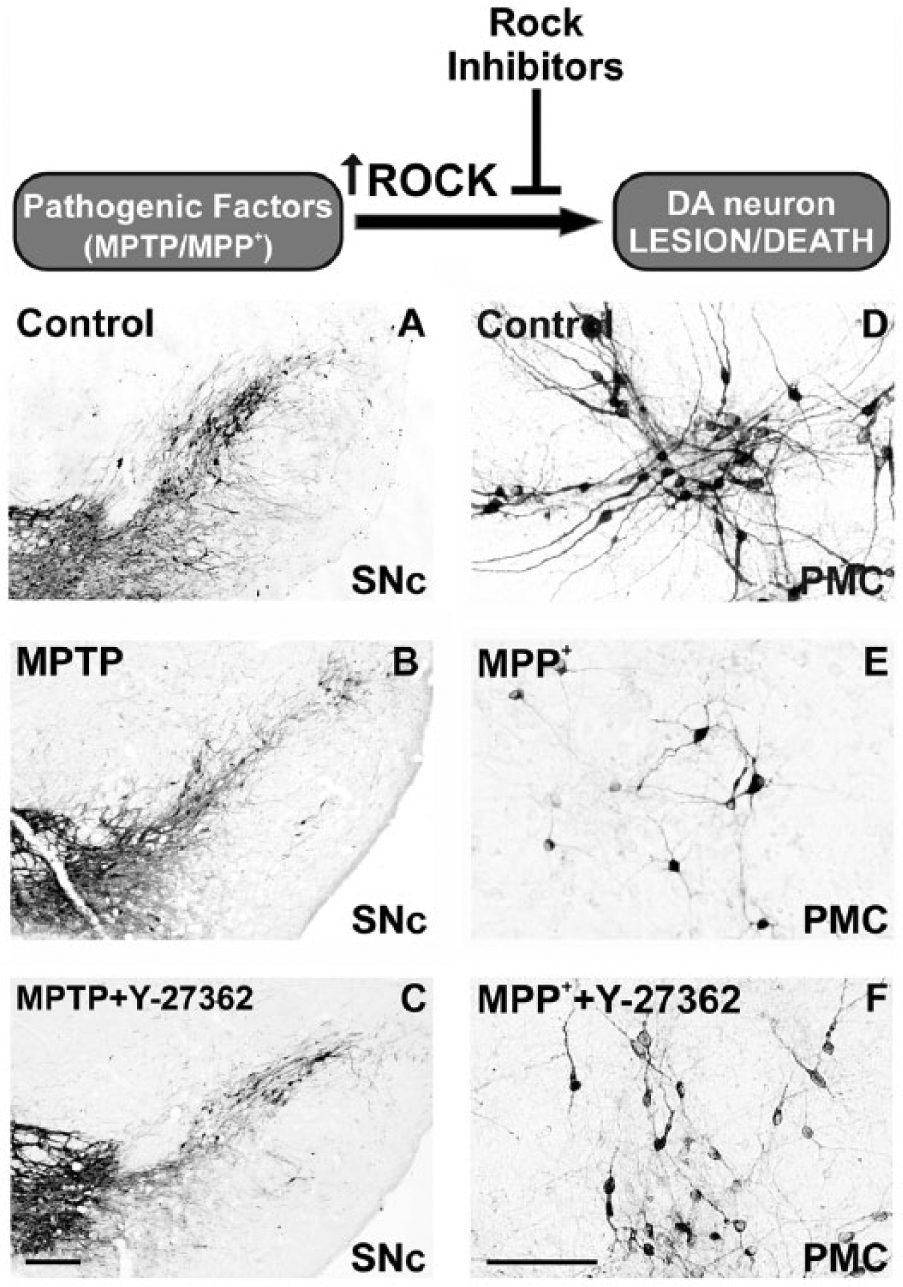
ROCK inhibition protects against dopaminergic cell death. In mice (A-C) and primary mesencephalic cultures (D-F), it induced at loss of dopaminergic neurons in the substantia nigra compacta and cultures (B, E) relative to controls (A, D), and this loss was significantly reduced by simultaneous treatment with the ROCK inhibitor Y-27632 (C, F). Scale bar: 250 µm (A-C) and 100 µm (D-F). DA = dopaminergic; PMC = primary mesencephalic cultures; SNc = substantia nigra compacta. For details, see Villar-Cheda and others (2012) and Rodriguez-Perez and others (2013).
Mechanisms Involved in ROCK-Induced Dopaminergic Vulnerability and Neuroprotective Effects of ROCK Inhibition
Several mechanisms appear to be involved in ROCK-induced dopaminergic vulnerability and the neuroprotective effects induced by ROCK inhibitors (Fig. 3). It has been suggested that ROCK inhibition activates neuroprotective survival cascades in dopaminergic neurons (Tönges and others 2012). A number of potential ROCK targets in apoptotic signaling have been suggested (for review, see Katoh and others 2014; Shi and Wei 2007). Interactions with Bcl-2 proteins, the pro-apoptotic factor glycogen synthase kinase 3 beta (GSK-3β) and Akt are particularly interestingly. The family of Bcl-2 proteins constitutes a group of crucial regulatory factors in apoptosis. Rho family G proteins regulate different proteins of the Bcl-2 family leading to changes in caspase 3 activity and apoptosis (He and others 2008). However, the effects of ROCK and ROCK inhibitors on Bcl-2 in different types of cells have been controversial (Hippenstiel and others 2002; Ikeda and others 2003; Koch and others 2014), and no data have been published on the effect of ROCK inhibition on Bcl-2 in PD models or dopaminergic neurons. A number of previous studies have suggested the association between GSK-3β and PD (Duka and others 2007; Hernandez-Baltazar and others 2013; Yu and others 2014), and GSK-3β inhibitors have been suggested as potential therapeutic agents for the treatment of PD (Morales-Garcia and others 2013). Interestingly, it has been observed that ROCK inhibitors also inactivated GSK-3β in several types of cells (Hamano and others 2012). However, controversial results have also been observed (Boku and others 2013), and the effect of ROCK on GSK-3β has not investigated in dopaminergic cells and PD models up to now. Previous studies have also shown that Akt activation induces trophic effects in murine models of PD (Ries and others 2006) and that phosphorylated Akt is depleted in brains of PD patients (Malagelada and others 2008). Furthermore, activation of the PI3K/Akt pathway led to inactivation of GSK-3β (Oster and others 2014). Interestingly, Akt signaling was significantly activated after ROCK inhibition with fasudil in primary mesencephalic cultures treated with MPP+ (Tönges and others 2012).
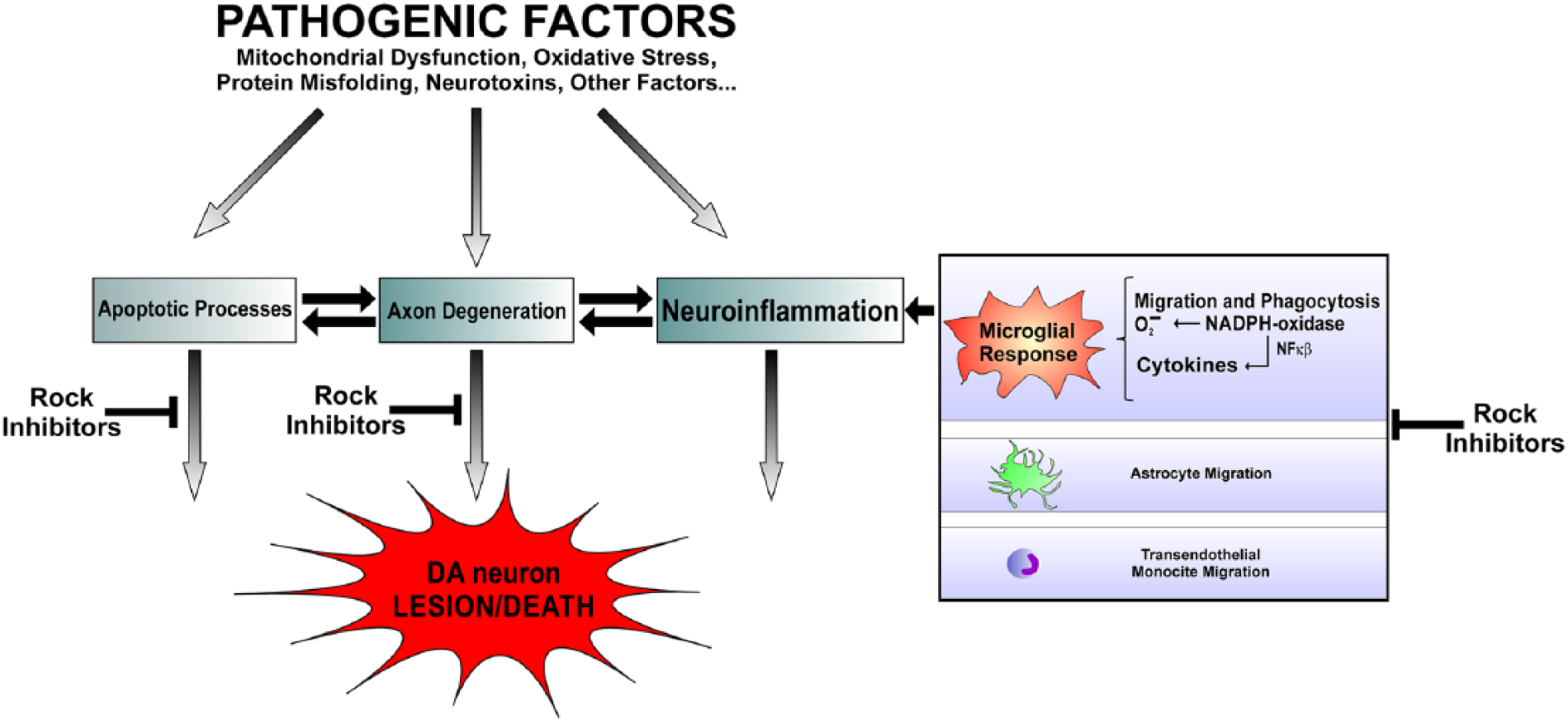
The pathogenic mechanism of Parkinson’s disease appears to be multifactorial. However, several major mechanisms involved in the progression of dopaminergic (DA) cell death such as apoptotic processes, axon degeneration, and neuroinflammation are related to ROCK activation and may be counteracted by ROCK inhibition. Glial cells, particularly microglial cells, play a crucial role in the neuroprotective effects of ROCK inhibition. ROCK has been involved in the microglial inflammatory response (migration, phagocytosis, NADPH-oxidase activation, production of oxidants, and inflammatory cytokines), as well as in migratory responses of astrocytic process and transendothelial migration of monocytes.
It has been suggested that AKt may also promote dopaminergic neuroprotection by inhibition of macroautophagy (Tönges and others 2012), because it has been observed that Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy (Cheng and others 2011) and that both pharmacologic and genetic disruption of autophagy signaling protected from axon degeneration (Yang and others 2007). Furthermore, it has been shown that the RhoA-ROCK pathway plays a key role in autophagy-associated suppression of axon elongation during the early stages of axonal growth (Ban and others 2013). However, it is also known that the disruption of autophagy in neurons is associated with progression of the pathology of several neurodegenerative diseases, including PD, Alzheimer’s disease, and Huntington’s disease (HD), and that macroautophagy is critical in the clearance of aggregated-prone proteins (Berger and others 2006; Dehay and others 2010). Furthermore, autophagy inducers have been suggested as potential tools in the treatment of PD and other proteinopathies (Crews and others 2010; Cuervo and others 2004). Interestingly, it has also been observed that inhibition of ROCK reduced the aggregation and levels of mutant huntingtin in cellular models of HD via activation of the ubiquitin proteasome system and macroautophagy and that this effect was mediated by ROCK I and ROCK II (Bauer and Nukina 2009; Bauer and others 2009). In addition, it has been observed that inhibition of Rho-GTPase signaling resulted in neurite extension and substantial reduction in expression of α-synuclein mRNA and protein in a dopaminergic cell line (Zhou and others 2011). In summary, it appears that ROCK activation is involved in the autophagic process, as autophagosome formation requires membrane remodeling to generate a vesicle. Afterwards, vesicles need to be transported in the cytoplasm, and the maturation of these vesicles involves fusion with other compartments. The actin cytoskeleton could potentially participate in each of these steps (Aguilera and others 2012). However, the consequences of RhoA-ROCK signaling in autophagy are poorly known, and possibly the cellular consequences of ROCK inhibition on autophagy can be deleterious or beneficial, depending on the precise context (Yang and others 2013).
Axon-stabilizing effects in damaged neurons may represent another mechanism of neuroprotection of dopaminergic neurons by ROCK inhibition (Borisoff and others 2003; Wakita and others 2014). In neurons, ROCK activation has been shown to be involved in axonal collapse and retraction in the presence of neurotoxins and other inhibitory conditions, through modulation of myosin light chain, phosphorylation of LIM kinase, and other mechanisms (Borisoff and others 2003; Gallo 2004; Lehmann and others 1999). Consistent with this, treatment of primary (neuron-glia) mesencephalic cultures with MPP+ led to a significant reduction in the length of the neurites of the surviving TH-ir neurons. However, simultaneous treatment with the ROCK inhibitors led to significant lengthening of the neurites of the surviving TH-ir neurons (i.e., significant decrease in axonal collapse and retraction; Borrajo and others 2014b; Tönges and others 2012). Interestingly, a similar reduction in the length of the neurites of the surviving TH-ir neurons was induced by MPP+ in cultures lacking microglia and cultures of the dopaminergic neuronal cell line MES 23.5, and treatment with the ROCK inhibitors also induced a significant increase in the length of dopaminergic neurites (Borrajo and others 2014b). This reveals a direct effect of ROCK inhibition on the dopaminergic neurons to stabilize neurite length rather than an indirect effect due to glia-derived mechanisms.
However, glial cells have been shown to play a crucial role in the neuroprotective effects of ROCK inhibition on dopaminergic neurons (Barcia and others 2012; Borrajo and others 2014b; Villar-Cheda and others 2012) and other neurons (Tönges and others 2014). In PD models, treatment with MPTP/MPP+ increased ROCK activity in mice and in primary neuron-glia cultures; however, no significant MPP+-induced increase in ROCK activity was detected in cultures lacking microglia; in addition, treatment with ROCK inhibitors blocked the MPTP-induced microglial activation in mice and primary mesencephalic cultures (Villar-Cheda and others 2012). Therefore, it was suggested that inhibition of microglial ROCK is essential for dopaminergic neuroprotection. The crucial role of microglial cells in the neuroprotective effects of ROCK inhibition on dopaminergic cell death was further demonstrated in a recent study by Borrajo and others (2014b). In primary (neuron-glia) cultures, simultaneous treatment with MPP+ and the ROCK inhibitor Y-27632 significantly reduced the loss of dopaminergic neurons. However, in the absence of microglia (primary cultures lacking microglia or cultures of the 23.5 dopaminergic neuron cell line), treatment with the ROCK inhibitor did not significantly reduce the dopaminergic cell loss. Inhibition of microglial ROCK activity is essential for the neuroprotective effects exerted by ROCK inhibitors against the dopaminergic cell death induced by MPTP/MPP+.
In addition to the essential role of microglia, other glial cells may also contribute to the neuroprotective effects of ROCK inhibition. In astrocytes, ROCK has been associated with the growth and migratory responses of astrocytic process (Höltje and others 2005; Lau and others 2011), and ROCK inhibitors have been observed to modulate astrocyte-derived axonal growth factors (Chan and others 2007), and granulocyte colony-stimulating factor production (Ding and others 2009). In oligodendrocytes, ROCK has been shown to be involved in control of polarity and directional migration to axons during developmental myelination and to injured sites in adults (Binamé and others 2013), as well as regulation of the levels of reactive oxygen species (ROS; Paintlia and others 2013). However, several studies have shown that ROCK expression is more abundant in microglia than in neurons and other glial cells (Ding and others 2010; Villar-Cheda and others 2012; Zhang and others 2008), and the above-mentioned studies show that inhibition of the microglial ROCK is essential for induction of a significant decrease in dopaminergic neuron death. Activation of microglial ROCK mediates at least 3 major components of the microglial inflammatory response. First, in inflammatory cells (Greenwood and others 2003; Honing and others 2004), including microglia (Barcia and others 2012; Bernhart and others 2010; Yan and others 2012), RhoA/ROCK is an important regulator of the actin cytoskeleton, which is particularly important for migration into inflamed areas and several changes involved in phagocytosis. In an interesting study, Barcia and others (2012) have shown that, in mice, MPTP treatment promotes polarization of microglial cells and formation of dopaminergic neuron-microglia contacts, which precede neuron elimination. However, ROCK inhibition blocked activating features of microglia such as increased cell size and number of filopodia. Second, ROCK interacts with NADPH oxidase (see below) and ROCK inhibitors suppress activation of NADPH oxidase (Budzyn and others 2006; Higashi and others 2003; Moon and others 2013). In microglia and other inflammatory cells, NADPH-oxidase produces high concentrations of ROS that are released extracellularly for killing invading microorganisms or cells (Babior 2004; Ray and Shah 2005). Finally, ROCK inhibition blocks microglial release of inflammatory cytokines such as interleukin-β and tumor necrosis factor-α (Borrajo and others 2014a; Zhang and others 2013).
Alteration of the cerebral microvascular endothelium, breakdown of the blood-brain barrier (BBB) and increased leukocyte transmigration have been involved in the pathogenesis of PD and other neurodegenerative diseases (Chung and others 2010; Grammas and others 2011). It has been suggested that absorption or metabolism of putative PD toxins and/or their faulty of elimination across the BBB has a role in the pathogenesis of PD. The possible damaging effects of pesticides and metal ions have been particularly investigated (Goldman 2014; Jellinger 2013; Mendez-Alvarez and others 2002). Furthermore, although the CNS has classically been considered an immunologically privileged region due to the presence of innate microglia, a number of studies suggest the potential role of infiltrated peripheral immune cells in degeneration of dopaminergic neurons in PD (Brochard and others 2009; Chung and others 2010). Enhancement of ROCK activity and ROCK inhibitors may affect neuroinflammation and dopaminergic degeneration by inducing changes in the permeability of brain vessels and infiltration of inflammatory cells. Several studies have shown that ROCK signaling is involved in microvascular endothelial hyperpermeability induced by thrombin (Carbajal and others 2000; Gavard and Gutkind 2008), VEGF (Bryan and others 2010; Sun and others 2006), and other factors (Breslin and others 2006; Chen and others 2011). ROCK is known to enhance an association between actin and myosin that leads to vascular smooth muscle contraction and vasoconstriction (Amano and others 1996). In addition, it has been observed that pericytes can regulate vascular basement membrane remodeling and govern neutrophil extravasation during inflammation by controlling the size of pericyte gaps, and that activated inflammatory cells bind to pericytes and initiate a signaling cascade that acts on RhoA/ROCK pathway in pericytes and modulates pericyte contractility and pericyte gaps (Wang and others 2012). Crosstalk between ROCK and endothelial function has also been suggested to be critical (Noma and others 2012).
The interaction between VEGF and ROCK is particularly interesting. Although low levels of VEGF may have neuroprotective effects on dopaminergic neurons (Yasuhara and others 2004), high doses of VEGF lead to poor circulation, brain edema, and disruption of BBB in PD models, which may contribute to dopaminergic degeneration (Barcia and others 2005; Muñoz and others 2014; Yasuhara and others 2005). Microvascular alterations and overexpression of VEGF and other markers of angiogenesis have also been observed in postmortem PD brains (Wada and others 2006; Yasuda and others 2007). It has been observed that VEGF-driven angiogenesis is largely mediated by ROCK II (Bryan and others 2010). ROCK signaling is an important mediator in a number of angiogenic processes including migration, survival, and permeability of endothelial cells. Consistent with this, it has been shown that ROCK inhibition blocked VEGF-induced microvascular endothelial hyperpermeability (Sun and others 2006), and ROCK inhibitors have been suggested as a useful tool for the treatment of angiogenesis-related disorders.
The role of ROCK in promotion of transendothelial migration and infiltration peripheral immune cells into the CNS is not only related to the above-mentioned effects on the microvascular permeability. ROCK activation in inflammatory cells may promote cell migration by regulating cytoskeletal dynamics and cell adhesion, as ROCK is active at the leading edge in lamellipodia and filopodia of crawling and transmigrating cells (Heasman and Ridley 2011; Honing and others 2004). Finally, it is important to note that the above-mentioned in vitro experiments in PD models (i.e., primary cultures of dopaminergic neurons) show that ROCK inhibition is able to induce important neuroprotective effects against dopaminergic neuron degeneration in the absence of any effect on the microvascular endothelial permeability, BBB function, or transendothelial migration of peripheral immune cells.
In summary, the pathogenic mechanism of PD appears to be multifactorial and ROCK is involved in a large number of cellular processes. Therefore, a number of the above-mentioned mechanisms may be involved in the effects of ROCK on dopaminergic neuron degeneration and the neuroprotective effects of ROCK inhibitors. However, only a few of these mechanisms have been confirmed in PD models up to now. Furthermore, it has shown that, in PD models, ROCK interacts with several factors that play a major role in dopaminergic neuron degeneration/neuroprotection such as NADPH-oxidase, angiotensin, and estrogen (Fig. 4).
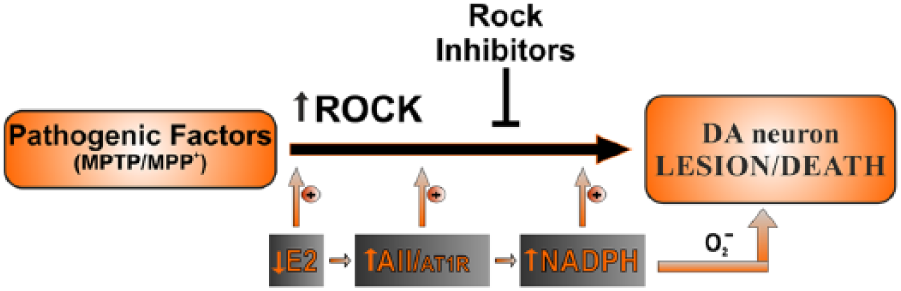
ROCK interacts with several brain factors that play a major role in dopaminergic neuron vulnerability such as NADPH-oxidase, angiotensin, and estrogen. Loss of estrogen, angiotensin/AT1 hyperactivity, and NADPH-oxidase activation increase ROCK activity and dopaminergic cell death. AII = angiotensin II; AT1 = angiotensin type 1 receptor; E2 = estrogen; NADPH = NADPH-oxidase; O2− = superoxide.
Interactions between ROCK and NADPH-Oxidase in Dopaminergic Cell Death
Several studies in peripheral noninflammatory cells, such as vascular cells, have suggested that there may be some interaction between ROCK and NADPH-oxidase, as ROCK inhibitors suppress NADPH oxidase activation (Budzyn and others 2006; Higashi and others 2003) and NADPH-oxidase inhibitors suppress ROCK activity (Li and others 2012). However, other studies suggest that ROCK may be activated independently of NADPH-oxidase (Hiroki and others 2004; Mattson and Maudsley 2009; Ohtsu and others 2006). This important issue must be clarified in microglia, and the corresponding experiments have already been carried out in our laboratory. We have observed that NADPH-oxidase, via NF-kB, activates ROCK, which, in turn, activates NADPH-oxidase. This is consistent with previous studies in peripheral cells suggesting that NADPH-oxidase-derived superoxide can activate NF-kB (Mattson and Maudsley 2009) and NF-kB activates ROCK (Hiroki and others 2004). Activation of the NADPH-oxidase complex is a key mechanism in the microglial response. Several experimental studies have indicated that microglial activation and induction of ROS by NADPH-oxidase constitute early components of DA cell death and that both factors act synergistically with other factors to induce DA cell death at early stages of the lesion process (Gao and others 2002; Gao and others 2003; Rodriguez-Pallares and others 2007; Wu and others 2003). The NADPH oxidase complex is the most important intracellular source of ROS other than mitochondria (Babior 1999, 2004). NADPH oxidase is a multicomponent enzyme composed of three cytosolic proteins or subunits and at least two membrane proteins. These components are spatially separated and the complex is inactive. The NADPH complex is assembled and activated after stimulation. The best-known NADPH oxidase is that of phagocytes-neutrophils and monocytes. In these cells and in microglial cells in the nervous system, the enzyme produces large quantities of extracellular superoxide and other reactive oxidants as part of the host defence system (Babior 1999, 2004). After phagocytosis in macrophages, there is an abrupt increase in superoxide formation, known as oxidative burst, which is catalyzed by the membrane NADPH-oxidase complex (Babior 1999). In addition, NADPH produces intracellular ROS that act on intracellular signaling pathways to induce microglial activation and amplify production of proinflammatory molecules (Qin and others 2004). More recent studies have shown that a number of different cell types (including neurons and astrocytes) contain NADPH oxidases. However, in contrast to the high production of oxidants by phagocytes and microglia, other cell types produce ROS at low rate and have a signaling function. It has been shown that only NADPH-oxidase from microglia, and not astrocytes and neurons, cause NADPH-oxidase-mediated neuron damage (Qin and others 2004). It has been suggested that NADPH-induced ROS may initially have developed as an intracellular signaling mechanism common to all cell types, and later as a specialized defense system in macrophage cells (Babior 1999, 2004).
Interactions between ROCK and Angiotensin in Dopaminergic Cell Death
The renin–angiotensin system (RAS) was initially recognized as a circulating humoral system involved in the regulation of blood pressure. It is now known that in addition to the circulating RAS, local (tissue or paracrine) RAS exist in many tissues (Re 2004), including brain tissue (Labandeira-Garcia and others 2012; Labandeira-Garcia and others 2013; Saavedra 2005). The actions of angiotensin II (AII), which is the most important effector peptide of the RAS, are mediated by two main cell receptors: AII type 1 and 2 (AT1 and AT2). Hyperactivation of local RAS has been associated with decreased longevity and age-related degenerative changes in a number of tissues (Benigni and others 2009; Cassis and others 2010; de Cavanagh and others 2011; Min and others 2009), because the local RAS mediates (via AT1 receptors and NADPH oxidase activation) oxidative stress and several key events in inflammatory processes (Marchesi and others 2008; Mehta and others 2007). NADPH oxidase and also AT1 and AT2 receptors have been located in dopaminergic neurons and nigral microglia and astrocytes (Garrido-Gil and others 2013; Grammatopoulos and others 2007; Joglar and others 2009; Rodriguez-Pallares and others 2008; Valenzuela and others 2010). It has been shown that the dopaminergic cell loss induced by DA neurotoxins is enhanced by AII via AT1 receptors. Activation of the AII/AT1 pathway exacerbates the microglial NADPH oxidase and the glial inflammatory response, which are inhibited by treatment with AT1 antagonists such as candesartan (Joglar and others 2009; Rey and others 2007; Rodriguez-Pallares and others 2008). In addition, several studies have shown that the brain RAS plays a major role in other brain inflammatory diseases (Lanz and others 2010; Stegbauer and others 2009).
In a recent study by Villar-Cheda and others (2012), administration of the ROCK inhibitor Y-27632 induced a significant decrease in microglial activation and dopaminergic cell death induced by MPTP in mice, as well as a significant decrease in the enhancing effect of AII/AT1 activation on the microglial response and dopaminergic cell death. Consistent with this, the MPTP-induced increase in the expression of RhoA and in ROCK II mRNA levels and ROCK activity in the mouse substantia nigra were inhibited by AT1 receptor deletion. In cultured cells, the enhancing effect of AII on MPP+-induced dopaminergic neuron death was also inhibited by the ROCK inhibitor Y-27632, suggesting an important interaction between the AII/AT1 and the RhoA/ROCK II pathways in MPTP-induced dopaminergic neuron death. This is consistent with the results of several studies in the vessel wall, which have suggested the involvement of ROCK in AII-induced inflammatory arteriosclerotic and coronary lesions (Hiroki and others 2004; Iida and others 2008). The above-mentioned results suggest that both NADPH and ROCK activation may play a critical role in the AII-induced inflammatory response, which is counteracted by ROCK inhibitors.
In addition, ROCK inhibition fosters neurite outgrowth (Borisoff and others 2003; Lehmann and others 1999) and has axon-stabilizing functions (Gallo 2004) as indicated above. Interestingly, it is also known that inhibition of AT1 receptors and activation of AT2 receptors promotes axonal outgrowth (Laflamme and others 1996; Meffert and others 1996), axonal regeneration (Lucius and others 1998; Reinecke and others 2003), and neuron differentiation, the latter of which has also been observed in dopaminergic neurons in our laboratory (Rodriguez-Pallares and others 2004). Together these findings also suggest a possible interaction between angiotensin and ROCK in promoting axonal outgrowth.
Interactions between ROCK and Estrogen in Dopaminergic Cell Death
The results of animal model studies and clinical evidence together suggest that estrogen exerts a neuroprotective effect against PD. Furthermore, a number of epidemiological studies have reported that both the incidence and prevalence of PD are higher in men than in premenopausal women of similar age. Controversial effects of estrogen replacement therapy (ERT) have been also reported (Popat and others 2005; Shulman 2002). However, the effects of the timing of postmenopausal ERT and the age of the women receiving the treatment may be major factors in these discrepancies (Ragonese and others 2006a; Ragonese and others 2006b). The mechanism by which estrogen protects dopaminergic neurons has not been clarified. Modulation of the glial neuroinflammatory response by estrogen is involved in the neuroprotective effects (Morale and others 2006; Suzuki and others 2007; Tripanichkul and others 2006; Vegeto and others 2008), as neuroinflammation and microglial activation play a major role in the progression of PD (see above). Other mechanisms such as direct antiapoptotic (Brendel and others 2013; Das and others 2011) or trophic (Campos and others 2012; Lopez-Martin and others 1999) effects of estrogen on dopaminergic neurons have also been suggested.
In several studies with male and female rats and mice treated with 6-hydroxydopamine or MPTP, we have shown that estrogen inhibits the neurotoxin-induced neuroinflammatory response and dopaminergic cell death and that inhibition of nigral RAS activity plays a major role in the anti-inflammatory and neuroprotective effect of estrogen (Rodriguez-Perez and others 2010; Rodriguez-Perez and others 2011; Rodriguez-Perez and others 2012). It has also been observed that the increase in dopaminergic cell death induced by estrogen depletion is inhibited by the ROCK inhibitor Y-27632 and that estrogen depletion induces a significant increase in ROCK activity, which is blocked by treatment with the AT1 receptor antagonist candesartan (Rodriguez-Perez and others 2013). Furthermore, MPTP induces a marked increase in ROCK activity and RhoA and ROCK II mRNA and protein expression in ovariectomized mice, and these effects of MPTP are significantly lower in ovariectomized mice treated with estrogen replacement and in ovariectomized AT1-null mice (Rodriguez-Perez and others 2013). Together the findings suggest that ROCK activation may play a major role in the enhanced vulnerability of dopaminergic neurons after estrogen depletion and that this effect is mediated by activation of the AII/AT1 pathway.
Additional mechanisms may contribute to the beneficial effects of estrogen. Estrogen has been found to induce axon outgrowth in several types of neurons, although the mechanism has not been clarified (Gollapudi and Oblinger 2001; Mérot and others 2009). We have shown that estrogen induces ROCK inhibition (Rodriguez-Perez and others 2013), decreases AT1 receptor expression, and increases AT2 receptor expression (Rodriguez-Perez and others 2010; Rodriguez-Perez and others 2012); these are possible mechanisms underlying the estrogen-induced outgrowth of axons. The use of AT1 antagonists and ROCK inhibitors may provide a new neuroprotective strategy for addressing the higher susceptibility and progression of PD in postmenopausal women. This is of particular interest because both types of drugs are currently used against vascular diseases in clinical practice and may circumvent the potential risks of ERT. They may be particularly useful for women in whom ERT may be ineffective, such as elderly women or women affected by a long-term lack of estrogen.
Conclusion
ROCK inhibition protect dopaminergic neurons, and inhibition of the microglial neuroinflammatory response plays a major role in the protection. Furthermore, ROCK inhibitors have axon-stabilizing functions that contribute to neuroprotection and regeneration of dopaminergic neurons. Other mechanisms may also contribute to the neuroprotective effects of ROCK inhibitors on dopaminergic neurons and remain to be clarified. As ROCK possess multiple substrates, modulation of ROCK activity will be useful for treatment of many diseases, including PD. Fasudil, the only clinically available ROCK inhibitor, inhibits both ROCK I and ROCK II and may also affect other protein kinases such as PKA. It is necessary to develop more potent and selective inhibitors, possibly against ROCK II in PD, to reduce side effects and enhance the efficacy, and with better BBB penetration ability compared to fasudil. Several directions are being considered for future development of ROCK inhibitors, such as ROCK II isoform-selective inhibitors, “one-compound-multitargets” fasudil derivatives, or combination therapy of fasudil derivatives and complementary drugs. This may be particularly interesting for PD because the pathogenic mechanism of PD is multifactorial. Over the past few years, significant progress has been made for the development and application of ROCK inhibitors (for review, see Chen and others 2013; Chen and others 2014; Feng and LoGrasso 2014).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by Spanish Ministry of Economy and Competitiveness (BFU2012-37087), Spanish Ministry of Health (RD12/0019/0020 and Ciberned), Galician Government (XUGA), and European Regional Development Fund (FEDER).